The era in which science could conquer cancer has arrived, and the old reputation of cancer as an incurable disease is beginning to fade away. Each year new advances in medical research and clinical care diminish the old myth that cancer is a disease too complicated to comprehend and difficult to cure. The road to understanding and controlling of cancer remains arduous still, but recent progress provides reasons for cautious optimism.
Cancer facts and myths
Cancer is a class of diseases in which cells multiply out of control, invade surrounding tissues, and spread to distant organs in a process called metastasis. Invasion and metastasis are key features that distinguish malignant tumors—cancer proper—from benign growths. Cancer can emerge in essentially any organ of the body, and at any age. In developed countries, cancer is responsible for about one quarter of all deaths, and is beginning to surpass cardiovascular disease as the leading cause of death (American Cancer Society 2008; Jemal et al. 2005). Yet, in spite of these sobering realities, the notion that cancer is an incurable disease should be viewed as an obsolete myth. Most cancers can be treated, many can be successfully managed, and some can be completely cured. Cure rates for some cancers approach 95% of cases, a better success rate than that of some infectious diseases and metabolic disorders.
Fundamentally, cancer is a genetic problem. It emerges from mutations and other pathological changes in the genome of a cell, leading this cell and its descendants to misbehave (Vogelstein and Kinzler 2004). These alterations may be inherited at conception, affecting every cell of the body, but are more commonly acquired by accident in a small number of cells in one or another tissue. In most types of cancer, the transformation of a normal cell into a cancerous one requires multiple mutations that collectively disable key mechanisms for cellular self-control (Figure 1). This accumulation of mutations may take decades, which is one reason that cancer incidence increases with age.
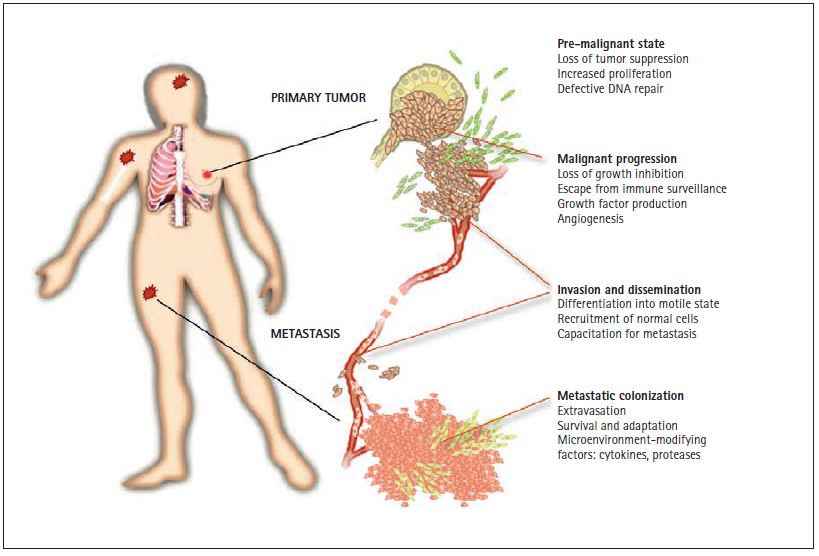
Figure 1. Phases of a solid tumor. Solid tumors such as carcinomas of the lung, colon, breast, or prostate start in epithelial cells that line the surface of the bronchia, the intestinal mucosa, or the alveoli of fl uid secretion in the breast and prostate. Mutations that increase the ability of these cells to proliferate generate small pre-malignant tissue masses. These pre-cancerous lesions may progress into malignant tumors by the acquisition of additional mutations that provide freedom from growth-inhibitory controls, protection from destruction by the immune system, capacity to invade the surrounding tissue, and the ability to attract blood capillaries (“angiogenesis”). A further conversion of the malignant tumor leads to the formation of highly motile and invasive cancer cells, and the recruitment of normal cells that act as helpers in tumor dissemination. These changes pave the way for the escape of cancer cells through the lymphatic system and the blood circulation to all parts of the body. Some disseminated cancer cells may have the ability to step out of the circulation (“extravasation”) by crossing the blood capillary walls. After they enter distant organs such as the bone marrow, the lungs, the liver, or the brain, cancer cells are able to survive, adapt, and eventually overtake these new environments, leading to the formation of lethal metastases.
Cancer is also a problem of cell biology. The genetic alterations that give rise to cancer act by disrupting the normal life cycle and social behavior of cells (Gupta and Massagué 2006; Hanahan and Weinberg 2000). Genes whose normal function is to promote cell movement and division may become cancer genes if they suffer alterations that increase these activities (Figure 2). On the other hand, genes whose normal function is to limit cell division, retain cells in place, promote cell differentiation, or eliminate spent and defective cells, lead to cancer when they suffer inactivation. The identification of cancer genes and the cellular functions that they control are at the forefront of contemporary research and anti-cancer drug development.
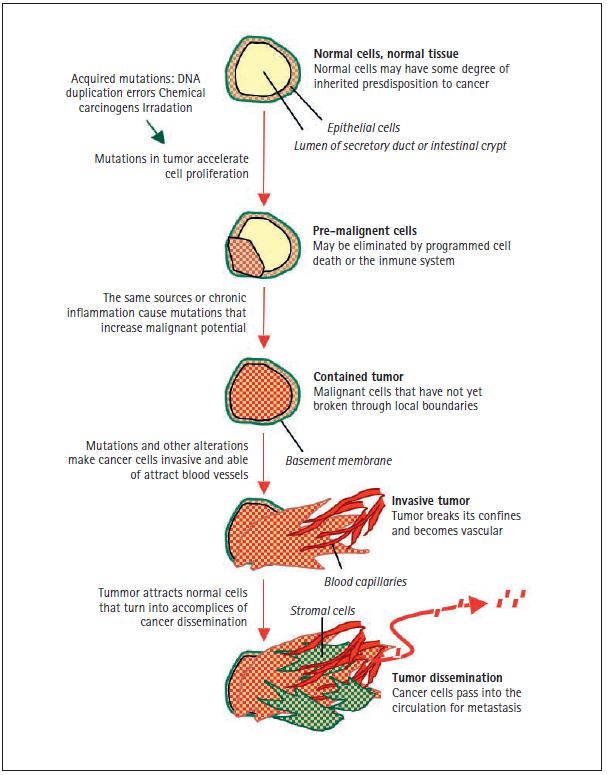
Figure 2. Sources of cancer mutations. The schematic represents the section of a secretory duct or an intestinal crypt, with a layer of epithelial cells surrounded by a basement membrane lining the cavity. The genetic inheritance of every individual contains a certain level of predisposition—high or low—for different types of cancer. Cancer-predisposing genetic variations that carry a small risk of developing a certain type of cancer are probably common in the human population, and most of these alterations remain to be discovered. Cancer-predisposing inherited mutations that carry a high risk of developing cancer (e.g. BRCA1 and BRCA2 mutations in breast and ovarian cancer, RB mutations in retinoblastoma, and APC mutations in colorectal carcinoma) are rare in the human population. These intrinsic cancer predispositions are present in all cells of the body. However, the initiation of tumor formation requires the acquisition of more mutations in all cases. The sources of cancer mutations may be internal, such as unrepaired errors in DNA replication that normal dividing cells make on their own, or external, such as chemical carcinogens in tobacco smoke or UV radiation in sun exposure. These acquired mutations accelerate cell proliferation and lead to the formation of pre-malignant lesions, such as intestinal polyps or breast tissue hyperplasias. Most of these lesions do not progress any further and are eliminated by cell self-destruction or by the killing action of the immune system. However, some pre-malignant lesions may progress into a contained carcinoma, or “carcinoma in situ” by the accumulation of additional mutations from external sources or from the genomic instability of the pre-cancerous cells. This stage is also facilitated by chronic inflammation syndromes that are triggered by a defective immune system (e.g. ulcerative colitis), an external irritant (e.g. tobacco smoke in the lungs), or an infectious agent (e.g., hepatitis virus in the liver, Helicobacter pilory in the stomach). A tumor becomes an invasive carcinoma when it breaks through the surrounding basement membrane and attracts blood capillary to bring in oxygen and nutrients. Epigenomic alterations in cancer cells, and stress in the surrounding tissue cause the release of factors that recruit normal cells, which end up being turned into helpers in tumor progression. At this stage the cancer cells have access to the circulation and can disseminate throughout the body. Some disseminated cancer cells may go on to reproduce the tumor in distant organs, giving rise to metastasis.
The identification of cancer genes and their biological functions during the last quarter of the 20th century is leading to better ways to prevent and treat cancer. Improved methods for assessment of cancer risk and more effective cancer prevention campaigns are decreasing cancer incidence and mortality in certain types of cancer. Less invasive surgical procedures, more refined radiotherapy methods, and more sophisticated use of chemotherapeutic drugs are contributing to the growing success of conventional cancer treatments. Moreover, a better understanding of biology and genetics of cancer is allowing the development of better drugs that target cancer cells while sparing healthy ones. And although these new drugs have begun to arrive at a trickle, this trickle is poised to become a flood. The achievement of these goals could be one of the principal scientific feats of the first half of the twenty-first century.
The growing incidence of cancer
Cancer is not a new disease. The Egyptians were surgically treating breast cancer circa 1600 BC (Karpozilos and Pavlidis 2004). By around 400 BC Hippocrates understood the difference between benign and malignant tumors, calling the latter “carcinoma” from the word “carcinos,” crab in Greek, referring to the shape that he saw in advanced malignant tumors, and “-oma” meaning swelling. But while cancer is not a new disease, its incidence is on the rise. Current estimates place the worldwide mortality from cancer at nearly 8 million people annually, or about 13% of all deaths (World Health Organization 2008). The World Health Organization forecasts that by 2020 the annual global death toll will rise to about 11.5 million.
Tumors result from the accumulation of multiple mutations in the affected cells. The multiple genetic changes that result in cancer can take many years to accumulate (Vogelstein and Kinzler 2004). This is why cancer in young children and adolescents is relatively rare, and why the risk of developing cancer increases with age. In developed countries an increase in life expectancy and in population median age over the past decades have contributed to an overall increase in cancer incidence. With progress in controlling infectious diseases that are currently ravaging underdeveloped countries, we may expect similar increases in cancer incidence in those countries as well. Other contributing factors include the detection of more early-stage tumors in routine medical examinations, various factors in diet and lifestyle, and the negative impact of cigarette smoking.
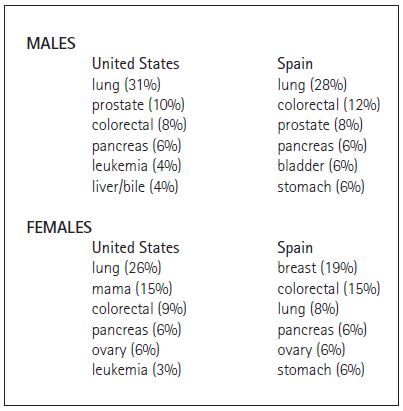
The overall incidence of cancer, and the incidence of particular types of cancer vary between different countries (Danaei et al. 2005; World Heatlh Organization 2008). For example, the most common types of cancer by mortality in the United States and Spain are similar, but with one notable exception: lung cancer mortality in females. Lung cancer ranks as the top cause of cancer deaths for males in both countries and for females in the United States. However, until recently at least, lung cancer is in third for position females in Spain (Table 1). This difference is attributed to the delay in tobacco consumption by women in Spain compared to men in both countries and women in the Unites States. Epidemiologic studies demonstrate a tight correlation between tobacco consumption and lung cancer, with a 20-year lag between the two.
Cancer and cancers
“Cancer” includes hundreds of different diseases. Primary tumors arising in different organs or tissues—for, example, breast cancer, lung cancer or leukemia—are obviously different in their appearance, evolution, response to treatment, and mortality. However, tumors arising in the same organ can be further classified into different subtypes that are very distinct from each other. There are at least five different subtypes of breast cancer, and even these could be subdivided into different variants. The same could be said of cancer in other organs. With these differences come distinct treatment indications.
Tumors are also classified according to the type of cell that they derive from. Carcinomas are malignant tumors derived from epithelial cells, such as those that form the upper layer of the skin and the digestive mucosa, or the internal structure of organs like the breast, prostate, liver, and pancreas. Sarcomas are derived from cells of connective tissues such as bone, cartilage, and muscle. Lymphomas and leukemias are derived from blood-forming cells, melanomas from melanocytes (skin pigmented cells), and glioblastoma, neuroblastoma, and medulloblastoma from immature neural tissue cells. Carcinomas are the most common type of cancer in adults whereas in the young neuroblastoma, medulloblastoma, and leukemia are the common types.
Table 1. Cancer incidence in adults in the United States (American Cancer Society 2008) and Spain (Centro Nacional de Epidemiología de España). Numbers in parenthesis represent the percent of all cancer deaths that are due to this particular type of cancer.
A third set of parameters in the classification of tumors is based on the extent of spread of the tumor, which is called “stage” of the disease, and the histological appearance under the microscope, which is called the “grade.” However, tumors of the same origin, kind, grade, and stage may progress and respond to therapy very differently in different patients. This reality has a major impact on our view of cancer as a disease that we still know too little about. Fortunately, this is about to radically change. The advent of molecular genetics technologies is allowing a better classification of cancers based on their specific origin, molecular alterations, risk of spread to other organs, and treatment of choice.
Causes of cancer
Cancer develops as a consequence of mutations and other abnormalities that alter the genes that control cell behavior (Hanahan and Weinberg 2000; Vogelstein and Kinzler 2004). These mutations may be acquired through the action of external factors—such chemical carcinogens, radiation and infectious agents—or internal errors in DNA replication and repair in small groups of cells throughout life (Figure 2). Cancer mutations may also be inherited, in which case they are in all cells from birth. Current research on the genetic basis of cancer is focusing on the processes that cause these genetic alterations, the types of genes that are affected, and the biological consequences of these effects.
Common examples of chemical carcinogens include tobacco smoking, which causes lung cancer and bladder cancer, and exposure to asbestos fibers, which causes mesothelioma (Danaei et al. 2005). Ultraviolet radiation from the sun can lead to melanoma and other skin cancer. Tobacco smoke carcinogens and radiation are thought to promote the formation of tumors by acting as direct mutagens. Tobacco and asbestos may also cause chronic inflammation that secondarily favors tumor development. Viral infections are the second most important external cause of cancer after tobacco usage (zur Hausen 1999). Viruses associated with human cancers include papilloma virus in cervical cancer, hepatitis B and C viruses in liver cancer, HIV in Kaposi’s sarcoma, and Epstein-Barr virus in B-cell lymphomas (Boshoff and Weiss 2002; Parato et al. 2005; Roden et al. 2006; Woodman et al. 2007; Young and Rickinson 2004). Viral infections promote tumor formation by incorporation of the virus genome into the DNA of the host cell, which may increase the activity of neighboring genes that stimulate uncontrolled cell division. Viral infections may also promote tumor growth by causing chronic inflammation and stimulating cell turnover in the host tissues. Liver tissue degeneration, or cirrhosis, caused by alcoholism, is associated with the development of liver cancer. The combination of cirrhosis and viral hepatitis constitutes the highest risk of developing liver cancer, which is one of the most common and deadly cancers worldwide. Certain bacterial infections also favor the development of cancer. The clearest example is gastric cancers tied to chronic inflammation of the stomach mucosa by Helicobacter pylori infection (Cheung et al. 2007; Wang et al. 2007).
Certain types of cancer have a strong hereditary component (Vogelstein and Kinzler 2004). Inherited mutations in the genes BRCA1 and BRCA2 create a high risk of developing breast cancer and ovarian cancer (Walsh and King 2007; Wang 2007; Welcsh and King 2001). Interestingly, BRCA mutations are rare in sporadic breast cancer. In contrast, p53, which is commonly mutated in sporadic cancers, is also the gene affected in the hereditary syndrome of Li-Fraumeni, which includes a predisposition for sarcomas, breast cancer, and brain tumors (Vousden and Lane 2007). Retinoblastoma in children is due to a hereditary mutation in the retinoblastoma (RB) gene, a gene which is also mutated in many sporadic cancers (Classon and Harlow 2002). An inherited mutation of the APC gene gives rise to thousands of polyps in the colon, which leads to early onset of colon carcinoma (Fodde et al. 2001). Another hereditary form of cancer predisposition is caused by mutations in one of several genes (MLH1, MSH2, MSH6, PMS1, PMS2) devoted to the repair of DNA replication errors. This condition, called HNPCC (hereditary non-polyposis colon cancer), include familial cases of colon cancer without a prevalence of colon polyps, uterine cancer, gastric cancer, and ovarian cancer (de la Chapelle 2004). Inherited mutations in the VHL1 gene predispose to kidney cancer (Kaelin 2005).
The inherited mutations that have a strong effect on tumor development are rare in the human population, and account for only a small fraction of cancer. For example, inherited BRCA mutations account for less than 2% of breast cancer in the general population (Welcsh and King 2001). At the other end of the spectrum, certain genetic variations may have a very weak individual impact on the risk of developing cancer but may be highly prevalent in the human population. In certain combinations, these genetic traits could synergize to create a significant risk of cancer. The current view is that cancer arises from complex interactions between external carcinogens and an individual’s genome. The identification of these weakly predisposing genetic determinants currently is a topic of intense investigation.
Normal cells and cancer cells
Cells are the basic unit of life. In isolation their basic activities are to resist the environment, incorporate nutrients, faithfully replicate their genome, and divide. However, the cells that form the tissues of a complex organism can no longer perform these tasks autonomously. Single cells evolved to form organized colonies hundreds of millions of years ago because this communal form of life proved advantageous in facing up to harsh environments. But this communal life also meant giving up certain degrees of freedom. For example, it was no longer suitable for a cell in the community to divide or move just as it wished. In our highly organized tissues, such decisions are subject to a complex network of cell-to-cell molecular signals. This form of dialog between cells has been developing and enriching itself over millions of years, and a good portion of our genome is entirely devoted to this task.
Cells communicate with each other by secreting molecules, generally in the form of small proteins known as hormones, growth factors, cytokines, or chemokines. These factors contact receptor proteins on the surface of target cells to activate “pathways,” which are sequences of biochemical reactions between signal-transducing proteins inside the cell (Bierie and Moses 2006; Bild et al. 2006; Christofori 2006; Ciardiello and Tortora 2008; Classon and Harlow 2002; Ferrara 2002; Fodde et al. 2001; Hanahan and Weinberg 2000; Karin 2006; Malumbres and Barbacid 2007; Massagué 2004, 2008; Olsson et al. 2006; Pouyssegur et al. 2006; Sweet-Cordero et al. 2005; Vousden and Lane 2007). The end result of this process are positive or negative changes in the ability of the cell to move, metabolize, grow, divide, differentiate, or die. Other proteins inside the cell sense the presence of errors and alterations in the DNA, and prompt their repair or else provoke the death of the cell. Loss of these important signaling and self-controlling functions results in cancer. Cancer cells disobey essential rules of life in community, increasingly misuse proliferative stimuli, and ignore the rules of moderation. Their interaction with their neighbors becomes openly antisocial. They avert the policing action of the immune system. Eventually, they break through the physical barriers that encapsulate the tumor, setting out on the march that will spread cancer cells throughout the body and metastasis.
The mutations that cause cancer precisely affect the genes whose products exert these critical control functions. The progressive accumulation of mutations turn normal cells into pre-malignant and eventually into fully malignant cells (Figure 2). These changes can be observed under the microscope. A malignant process may start with the presence of an excessive number of normal-looking cells, called a hyperplasia, and more specifically with a disordered accumulation of such cells, or dysplasia. As the cells cease to look normal, the lesion is considered a carcinoma in situ, in which the abnormal cells are still confined to the normal limits of the tissue boundaries. When carcinoma cells invade the surrounding tissue by breaking through their underlying barrier (called the “basement membrane”), the lesion is call an invasive carcinoma. Each of these steps is accompanied by, and the result of, the progressively accumulating mutations that lead to cancer.
The specific functions that must be perturbed in order to generate cancer cells include a gain of self-sufficiency in growth-promoting signals; a loss of sensitivity to growth-inhibitory signals; a loss in the ability to undergo cell death (loss of apoptosis); a gain in the ability to perpetually replicate the DNA; and, a gain in the ability to evade surveillance by the immune system (Hanahan and Weinberg 2000). These changes are required for all types of cancer cells, including blood cell cancers such as leukemias. To form a tumor, cancer cells from solid tissues additionally require a gain in the ability to resist hypoxia through the induction of new capillaries that will feed the tumor (angiogenesis); and gain in the ability to detach and invade surrounding tissue (Figure 2). To spread the tumor to distant sites, the cancer cells must also gain the ability to pass into the circulation, enter distant tissues, and adapt to the microenvironment of those tissues eventually overtaking them.
Cancer genes
Cancer genes are divided into two general classes. Genes whose excessive activity contributes to cancer are called “oncogenes” (Hanahan and Weinberg 2000). The genes encode growth factor receptors such as EGFR and HER2, transducers of growth factor signals such as RAS, RAF, and PI3K, cell survival factors such as BCL2, and others. The mutations affecting these genes are activating or “gain-of-function” mutations. Genes whose normal activity prevents the emergence of cancer are called “tumor suppressor” genes. The mutations that affect these genes in cancer are inactivating mutations. Tumor suppressors include sensors of DNA damage such as p53, genes that fix DNA damage such as BRCA1 and BRCA2, inhibitors of the cell division cycle such as RB, receptors and transducers of growth inhibitory signals such as TGFBR and SMAD4, and suppressors of growth signals such as PTEN.
The mutations affecting these genes may be point mutations that alter one single nucleotide in the gene, and a single amino acid in the gene product. Point mutations may either increase or decrease the activity of the gene product, and so point mutations are a cause of oncogene activation as well as tumor suppressor gene inactivation. Small deletions or insertions may similarly cause either oncogene activation or tumor suppressor inactivation. Large-scale mutations involve the deletion or gain of a portion of a chromosome, resulting in the gain of multiple copies of one or more oncogenes, or a loss of tumor suppressor genes. Translocations occur when two separate chromosomal regions become abnormally fused, often at a characteristic location. A well-known example of this is the Philadelphia chromosome, or translocation of chromosomes 9 and 22, which occurs in chronic myelogenous leukemia, and results in production of the BCR-ABL fusion protein (Melo and Barnes 2007). This causes oncogenic activation of the ABL gene. Some oncogenic mutations affect not the protein-coding region of an oncogene but the regulatory or “promoter” region that controls the production of the gene product. Insertion of viral genome near the promoter region may also lead to hyperactivation of an oncogene.
In addition to the various types of mutations that alter the chemical structure of a normal gene turning it into a cancer gene, there is a growing recognition of the impact of epigenomic modifications. These are chemical modifications of the DNA and the proteins that envelop it (Blasco 2007; Esteller 2007). These modifications are known as “epigenetic” changes and can either render a gene silent or make it competent for activation. Epigenetic deregulation can contribute to cancer by failing to keep oncogenes silent, for example, through DNA methylation. Loss of methylation can induce the aberrant expression of oncogenes. Methylation or acetylation of histone proteins that package the chromosomal DNA can also suffer alterations that contribute to cancer. The experimental anti-cancer drug vorinostat acts by restoring histone acetylation and is currently undergoing clinical evaluation.
Ecology of the tumor microenvironment
Each tissue has a characteristic structure, boundaries, vascular supply, and extracellular milieu of hormones, nutrients, and metabolites. Cancer cells that alter this become exposed to environmental stresses including lack of oxygen (hypoxia) and nutrients, acidity, oxidative stress, and inflammatory responses. These stressful conditions select for cells that survive such pressures and become a dominant population in the growing tumor. This phenomenon is known as “clonal selection” (Nowell 1976). The resulting clones of cells are not merely survivors; they are highly effective profiteers that take advantage of the tumor microenvironment.
Tumors are more than just a conglomerate of cancer cells. Tumors also include normal cells that become attracted to, and engulfed by the growing tumor, and may become accomplices in its development (Joyce 2005; Mueller and Fusenig 2004). The collection of non-cancerous cell types that are present in a tumor is called the tumor “stroma,” and their importance in cancer is being increasingly recognized. Endothelial cells recruited into the tumor form new blood capillaries that bring nutrients and oxygen into the tumor mass. Macrophages and other immune and inflammatory cells congregate in the tumor in an attempt to respond to the tissue distress. Tumor-associated macrophages, produce growth factors and ECM-degrading enzymes that stimulate the growth and invasion of the cancer cells (Joyce 2005; Lewis and Pollard 2006). Stress-response cells are also recruited into the tumor from the circulation. Several types of blood-derived cells are attracted by signals that emanate from the tumor and proliferate in response to these signals. Stroma-derived factors may in turn stimulate cancer cells to release signals that enhance their ability for form metastases. For example, the stroma-derived cytokine transforming growth factor ? (TGF?) can induce breast cancer cells to release angiopoietin-like 4, which enhances the ability of these cells to seed the lungs after they escape from the primary tumor (Padua et al. 2008). Thus, the stroma of a tumor can provide cancer cells with metastatic advantages.
Metastasis: the deadly spread of tumors
Aggressive tumors may release millions of cancer cells into the circulation before the tumor is detected and surgically removed. Metastasis is the process by which these disseminated cancer cells take over distant organs and ultimately cause organ dysfunction and death (Figure 1). Metastases may be detected at the time of initial diagnosis of cancer or months to years later, when the disease recurs. The disseminated cancer cells may remain dormant in distant organs for a long period, until unknown conditions lead to their reactivation and formation of aggressively growing metastasis.
The administration of chemotherapy to cancer patients after surgical removal of a primary tumor is intended to eliminate all residual tumor cells and avert the eventual emergence of metastasis. Yet, the failure of current therapeutics to control or cure metastasis is responsible for 90% of cancer deaths. If it were not for metastasis, cancer would represent only a small fraction of the problem that it is today. Understanding the many molecular players and processes involved in metastasis may eventually lead to more effective, targeted approaches to treat cancer.
Recent advances in technologies to visualize and track the metastasis process have helped delineate multiple events that lead cancer cells in a primary tumor to reach and colonize a distant site (Fidler 2003; Gupta and Massagué 2006; Weinberg 2007) (Figure 2). Carcinoma cells must first pass through the basement membrane of the tissue compartment in which the tumor occurs. The basement membrane separates the epithelial cell layers in which carcinomas originate, from the subjacent tissue. Basement membranes also envelop the blood vessels. In order to surpass a basement membrane and spread through the surrounding tissue, cancer cells must acquire the ability to detach from their place of origin, adopt a migratory behavior, and release proteolytic enzymes that degrade the protein scaffold of the basement membrane and extracellular matrix.
Once cancer cells form a small tumor mass and create hypoxic conditions, they respond to hypoxia with the secretion of cytokines that stimulate the formation of new capillaries that bring in oxygen and nutrients for tumor growth. As a result of tumor-derived permeability factors, these newly formed capillaries are leaky, providing a route for the escape of the cancer cells into the blood circulation. Lymphatic vessels that drain fluid from the tumor and surrounding tissue provide another route for cancer cell dissemination. Lymph nodes frequently trap traveling tumor cells and document their spread, which is why lymph node status is an important prognostic indicator at initial diagnosis. However, the dissemination of cancer cells to distant organs such as the lungs, brain, bones, and liver occurs mainly through the blood circulation. In the bloodstream, cancer cells associate with each other and blood cells to form emboli that may help withstand mechanical stresses and evade surveillance by the immune system.
Once cancer cells lodge in capillaries at distant organs they must pass through the capillary walls in order to gain access to the organ parenchyma (Figure 3). Extravasation, as this process is known, depends on the ability of the cancer cells to disrupt the tight contacts between endothelial cells of the capillary wall and the enveloping basement membrane. The microenvironment of the infiltrated organ is largely not permissive for the extravasating cancer cells, many of which die. Those that survive form micrometastases that must adapt to the new environment and co-opt its resident cells in order to re-initiate tumor growth and form aggressive metastatic colonies. This process can take months, years, and even decades. Only a small fraction of the cancer cells released by a tumor are capable of fulfilling all these requirements, but the few that do are sufficient for the establishment of lethal metastases.
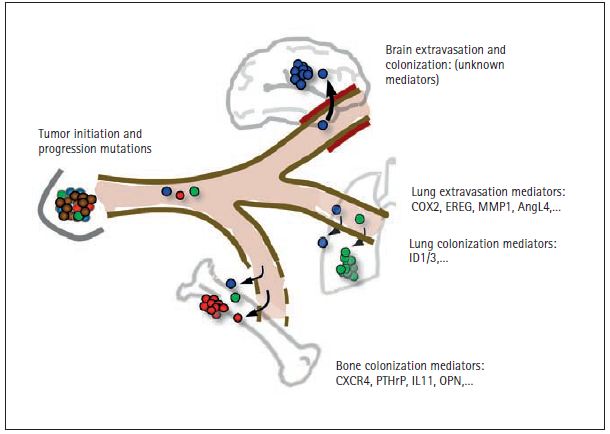
Figure 3. Mediators of distant metastasis in breast cancer. Metastasis has been most extensively studied in breast cancer because of the common nature of this disease, the availability of clinical material, and the characteristic set of organs that are affected in this process. Breast tumor can release cancer cells into the circulation as soon as they become locally invasive through the acquisition of tumor initiating and tumor progression mutations (see Figure 2). The disseminated cells that survive the physical stresses of the circulation require additional functions for entry into distant tissues. The passage through the blood capillary walls in those tissues, or “extravasation,” is relatively permissive in the bone marrow (and in the liver, not illustrated), because the capillaries in these tissues have natural windows for the constant entry and exit of blood cells. However, on entering the bone marrow, cancer cells must have the ability to survive and productively interact with this microenvironment. The fact that metastasis from breast cancer may take years and even decades to emerge suggests that the disseminated cancer cells originally arrived in this organ unprepared, and they had to slowly evolve the necessary abilities to expand as aggressive colonies. The genes that breast cancer cells misuse in order to survive in the bone marrow include the chemotaxis and survival receptor CXCR4, the osteoclast stimulating factors parathyroid hormone-relaped protein (PTHrP), interleukin-11 (IL11) and osteopontin (OPN), and other genes. In contrast to the bone marrow capillaries, the capillaries in other organs such as the lungs and especially the brain have tight walls that are very restrictive to the passage of circulating cells. For this reason, cancer cells must carry certain activated genes in order to enter these organs. Mediators of breast cancer cell entry into the lungs include the EGFR ligand epiregulin (EREG), the prostaglandin-synthesizing enzyme cycloxygenase-2 (COX2), the collagen-degrading enzyme matrix metalloproteinase-1 (MMP1), and the endothelium disrupting factor angiopoietin-like 4 (AngL4). It is suspected that some of these genes are also engaged by breast cancer cells to enter the brain. The genes that mediate colonization of the lungs and the brain are largely unknown, and the subject of active investigation. ID1 and ID3 have been recently identifi ed as mediators of tumor re-initiation by breast cancer cells entering the lungs. Thus, expression of ID1/3 is a property of tumor-propagating cells, also known as “cancer stem cells”.
The ingredients for metastasis Genetic heterogeneity
Metastasis has many features of a Darwinian evolution process in which selective pressures for the emergence of the fittest individual cells from a tumor cell population. Evolution requires the presence of genetic heterogeneity in a population from which fit individuals can be selected to match particular environmental pressures. In tumors, such heterogeneity is amply provided by the characteristic genomic instability of cancer cells, and it increases the probability that some cells in a tumor will achieve metastatic competence. Thus, the different steps of metastasis do not necessarily represent the acquisition of individual specialized mutations but rather represent the random accumulation of traits that provide the necessary advantage for adaptation to a different organ microenvironent.
Genomic instability and heterogeneity of cancer cells are apparent in the chromosomal gains, losses, and rearrangements found in tumors. DNA integrity can be compromised by aberrant cell cycle progression, telomeric crisis, inactivation of DNA repair genes, and altered epigenetic control mechanisms. For example, one half of all human cancers suffer loss of the tumor suppressor p53, an internal protein that responds to DNA damage by causing elimination of the damaged cell. The loss of p53 allows cancer cells with DNA alterations to survive and accumulate additional mutations (Halazonetis et al. 2008). Inherited mutations in certain DNA repair genes are associated with a higher risk of developing cancer, for example, in the hereditary nonpolyposis colorectal cancer syndrome (HNPCC) (Rustgi 2007), and in familial breast cancer syndromes cause by mutations in BRCA1 or BRCA2 (Martin et al. 2008).
Cancer stem cells
Metastasis requires a robust ability of cancer cells to re-initiate tumor growth after they penetrate a distant tissue in small numbers. Not all the cancer cells in a tumor are capable of dividing indefinitely and so not all cancer cells have the capacity to re-initiate a tumor after they arrive in a metastasis site. Many in fact loose tumorigenic power by falling into a more differentiated state. However, by one mechanism or another a subset of cancer cells in a tumor have the capacity of acting as tumor-propagating cells (Clarke and Fuller 2006). This capacity defines the cells that have it as “cancer stem cells” or “tumor-propagating cells.” These functions would support the maintenance of primary tumors, and would be essential for the establishment of metastatic colonies. Additional properties of these cells may include resistance to chemotherapeutic drugs or sensitivity to different drugs compared to the sensitivity of the bulk cancer cell population in the tumor. However, tumor propagating cells need not be a minority of the cancer cells in a tumor; in some types of tumors a majority of the cells may be competent to re-initiate tumor growth if they fulfill the other requirements for metastasis. The extent to which different tumor types may be initiated and sustained by cancer cells that meet these criteria is a subject of intense investigation and one that is likely to meet with different answers in different tumor types.
Metastatic dissemination
In order to become disseminated, cancer cells must be able to break their ties with the cohesive structure of the tissue of origin. Adhesion of cancer cells to each other is reduced by the loss of cell-cell anchoring proteins such as E-cadherin. Loss of E-cadherin in tumors can occur through various mechanisms, including silencing of the gene that encodes E-cadherin, mutations in this gene that result in the production of inactive E-cadherin, or repression of E-cadherin activity by growth factor receptors (Perl et al. 1998; Thiery 2002). Loss of E-cadherin activity also occurs as part of the transformation of cancer cells from an epithelial state into a more motile cell state, a change known as “epithelial-mesenchymal transition” or EMT (Cano et al. 2000; Yang et al. 2004). Normal cells are also kept in place by the extracellular matrix (ECM), a scaffold formed by collagen and other fiber-forming proteins to which cells attach by means of receptors called integrins. These ECM contacts can retain cells in place but can also stimulate cells to form extensions for migratory movement. Various proteins involved in these types of cell shape changes, such as RhoC and NEDD9, have been implicated in cancer cell invasion leading to metastasis (Clark et al. 2000; Kim et al. 2006).
Cancer cells may disseminate from a tumor very early on in a tumor formation process. Cancer cells have been detected in the bone marrow of breast cancer patients with small tumors (Klein et al. 2002; Schmidt-Kittler et al. 2003). This does not necessarily mean that the earliest departing cells are the ones that progress into overt metastasis, but it does indicate that dissemination is not an exclusive property of large, advanced tumors.
Once the disseminated cancer cells reach distant organs, they may remain dormant or die. Dormancy may last years, even decades before disseminated cancer cells burst into aggressive outgrowth, as in the case of breast cancer. Disseminated cancer cells found in the bone marrow of women or transgenic mice with early-stage breast cancer can become activated by transplantation into the bone marrow of mice to cause lethal tumors (Husemann et al. 2008).
Dissemination may also occur from metastatic tumors, which in turn seed new metastases. It is possible that circulating tumor cells can re-infiltrate the same tumors from which they departed. According to this hypothesis, tumors may continuously enrich themselves with their most aggressive progeny, providing a mechanism that couples metastatic ability with tumor growth (Norton and Massagué 2006). This would provide an explanation for the longstanding correlation between metastasis and tumor size (Minn et al. 2007). The timing and mechanisms of cancer cell dissemination are topics of great interest in contemporary cancer research.
Different “seeds” for different “soils”
The bones, lungs, liver, and brain are the most frequent sites of metastasis (Figure 3). However, different cancers have different proclivities to spread to these organs. (Billingsley et al. 1999; Gavrilovic and Posner 2005; Hess et al. 2006; Leiter et al. 2004). The compatibility between disseminated cancer cells (the “seed”) and certain distant organs (the “soil”) was already noted in the nineteenth century by Stephen Paget, who promulgated the “seed” and “soil” hypothesis (Paget 1889). For example, breast cancer can spread to these four sites, with bones and lungs being the most frequently affected. Lung cancer metastasis occurs with preference in the brain, bones, and contralateral lung. In contrast, prostate cancer metastasis principally occurs in the bones, and to a limited extent in the lungs. Furthermore, although these three tumors spread to the bones, they form very different types of lesions: bone metastasis from breast cancer and lung cancer is “osteolytic,” meaning that these lesions dissolve the bone matrix causing fractures. In contrast, prostate cancer metastasis is osteoblastic, leading to the generation of abnormal bone tissue that fills the marrow cavity. The predilection for a tumor in a particular organ to metastasize to another location in the same organ also varies. Tumors in one lung can easily metastasize to the other lung, whereas tumors in one breast rarely metastasize to the other breast.
Towards understanding metastasis
The progress achieved since the turn of the twenty-first century is shaping our view of metastasis based on a better understanding of its genetic, molecular, and biological bases. This knowledge is rapidly accumulating based on the identification of genes whose aberrant activity serves the purposes of the metastatic cells. Through these advances, metastasis is transforming from an obscure topic into a problem that is being rationalized and dissected, and may eventually be controlled.
An integrated model of metastasis
Early theories of metastasis proposed competing models of genetic predetermination of an entire tumor mass for metastasis, versus tumor progression giving rise to rare cells capable of metastasis (Vogelstein et al. 1988). With the sequencing of the human genome, powerful microarray technologies have been developed that allow researchers to determine the activation state of every gene in a small tissue sample. Using these techniques, it has been possible to identify patterns of gene activity, or “gene-expression signatures,” that can indicate the likelihood that a particular tumor will cause metastasis. If a sample extracted from a primary tumor shows the presence of a particular pro-metastatic gene-expression profile, this would indicate that a substantial proportion of the cells in that tumor are expressing such genes and thus are competent for metastasis. This would support the predetermination theory of metastasis. However, this competence may be quite incomplete. Additional alterations have to occur before the cancer cells become fully equipped to invade and colonize a distant tissue. The acquisition of a complete set of metastatic capacities may occur frequently in a tumor population, as must be the case in tumors that rapidly metastasize to multiple organs, or it may occur slowly in a minority of predisposed cells giving rise to metastases in one or another organ years or decades after departing from the primary tumor. The latter would argue for further progression of a tumor as a necessary step for metastasis.
Recent progress in metastasis research has provided experimental and clinical evidence for both the pre-determination and the progression models, leading to a model that integrates features of both. Cancer cells in a tumor with poor prognosis may contain activated genes that provide these cells with some, but not all the functions required for distant metastasis. We call these genes “metastasis progression” genes, because they directly allow the cancer cell population to become competent for metastatic behavior. Metastasis progression genes are necessary but not sufficient for metastatic outgrowth, because a majority of the cancer cells that express these genes are still incapable of forming metastatic tumors. This implies the existence of a complementary set of metastasis genes that provide additional survival and adaptation functions in a given organ. We refer to this class of genes as “metastasis virulence” genes.
Metastasis progression genes
Recent work in our laboratory has identified a set of 18 genes that breast cancer cells use to their advantage both in the primary tumor and in the lungs (Figure 3). This set, termed the “lung metastasis gene-expression signature” (LMS), includes EREG, COX-2, and MMP1, which cooperate in remodeling new blood capillaries in mammary tumors and existing lung capillaries when cancer cells expressing these genes reach the lungs. In mammary tumors the products of these genes support the assembly of leaky capillaries that facilitate the escape of cancer cells; in the lung, the same products facilitate the passage of circulating cancer cells into the parenchyma (Gupta et al. 2007). Another example is the gene that encodes ID1, which inhibits cell differentiation and stabilizes the tumor-propagating ability of cancer cells. In experimental models ID1 is important for the growth of breast tumors and for the re-initiation of the tumor growth after cancer cells reach the lungs. Thus, metastasis progression genes may couple the tissue-specific requirements of the microenvironment in a particular organ to a matching role in primary tumor progression. Breast cancer patients with LMS-positive primary tumors have a higher risk for developing lung metastases, but not metastases in bone or other sites.
Not all metastasis genes that are expressed in primary tumors provide a selective advantage in these tumors. For example, the production of transforming growth factor (TGF) in the stroma of breast primary tumors stimulates the expression of more than one-hundred genes in the breast cancer cells of the same tumor. Among these is the gene encoding the secreted factor ANGPTL4. Unlike EGFR, COX2, MMP1, or ID1, production of ANGPTL4 does not appear to provide an advantage to the cancer cells in the primary tumors—it merely reflects the presence of TGF in the tumor milieu. However, when the stimulated cancer cells reach the lung capillaries, the ANGPTL4 that these cells release causes disruption of the capillary walls and facilitates cancer cell entry into the tissue (Padua et al. 2008).
Specialized contributions of metastasis virulence genes
When cancer cells reach distant organs they are generally faced with a non-permissive microenvironment. To form a metastatic colony cancer must have an ability to resist and exploit this microenvironment. A clear example is provided by osteolytic bone metastasis from breast cancer. Circulating breast cancer cells that enter the bone marrow must find ways to survive in the unique stroma and hormonal milieu of this tissue, and ways to activate the mobilization and action of osteoclasts that mediate bone destruction. Breast cancer cells that are metastatic to bone express high levels of CXCR4. This membrane protein acts as the receptor for the cell survival factor CXCL12, which is abundantly produced in the bone marrow stroma (Wang et al. 2006). Therefore, cancer cells expressing high levels of CXCR4 obtain a specific advantage from the presence of CXCL12 in the bone marrow. In experimental models using mice, breast cancer cells that preferentially colonize the bones show not only high expression of the survival receptor CXCR4 but also an elevated production of the factors PTHrP (parathyroid hormone-related peptide), TNF-?, IL-1, IL-6, and IL-11 (Kang et al. 2003). When secreted by bone metastatic cells, these factors stimulate osteoblasts to release RANKL, which activates osteoclast differentiation. Osteoclasts dissolve bone, in turn releasing growth factors such as insulin-like growth factor-I (IGF-1), which favor cancer cell survival, and TGF?, which stimulates the cancer cells to further release PTHrP. The end result of this process is a vicious cycle of cancer cell-osteoclasts interactions that accelerated the destructive action of bone metastasis.
The ongoing search for genes and functions that mediate metastasis by other tumor types or to other organs is beginning to yield results. Prostate cancer cells secrete factors such as Wnt and bone morphogenetic proteins (BMPs) that stimulate the accumulation of osteoblasts. As a result, prostate cancer gives rise to osteoblasting (bone-forming) metastases, in contrast to the bone-destroying metastases caused by breast cancer. Compared to metastasis in bone and lung, little is known about the genes that cancer cells use to colonize the liver or the brain. However, this topic is under intense investigation and may yield progress in the near future.
Frontiers in cancer prevention, diagnosis, and treatment
Cancer prevention campaigns aiming at reducing high-risk behaviors (tobacco and alcohol abuse, sun exposure, and others) and routine screening for the detection of early-stage tumors are critical to reducing the incidence and mortality of cancer. Early diagnosis leads to therapeutic intervention before a tumor has become disseminated, curing the disease or at least extending the life of the patient. Important benefits have been obtained from screening for breast cancer with mammograms, colorectal cancer with colonoscopy, uterine cancer with cervical cytological testing, and prostate cancer with rectal exam and prostate-specific antigen (PSA) blood testing. Preventive vaccination against certain sexually transmitted strains of human papillomavirus is intended to reduce the incidence of cervical cancer. Genetic testing for certain cancer-related genetic mutations in BRCA1 and BRCA2 (which predispose to breast and ovarian cancers) and DNA repair genes (which predispose to colon cancer and other cancers) is performed in high-risk individuals with a family history of these diseases. Carriers of these mutations are subjected to close surveillance and may elect prophylactic surgery (removal of breasts, ovary, or colon) to reduce the risk of tumor development.
Recent progress is improving the classical approaches for the treatment of cancer (surgery, chemotherapy, radiation therapy) and novel approaches based on targeted therapy and immunotherapy. Surgical methods are gaining in precision and becoming less invasive. The surgical removal of a tumor that has not spread can effectively cure cancer. However, the propensity of cancer cells to invade adjacent tissue and spread to distant sites limits the effectiveness of surgery. Even small, localized tumors have metastatic potential. Therefore, surgery is very frequently complemented with other forms of therapy. Radiation therapy is based on the use of ionizing radiation (X-rays) to shrink tumors before surgery to kill locally disseminated cancer cells. Radiation can cause damage to normal tissue, therefore it can only be applied to a restricted area of the body. Radiation therapy destroys cells by causing extensive damage to their DNA. Most normal cells can recover from radiotherapy more efficiently than cancer cells, providing a window of opportunity for this intervention.
Chemotherapy is the treatment of cancer with drugs more toxic to cancer cells than they are to normal cells. Conventional anticancer drugs poison rapidly dividing cells by disrupting the duplication of DNA or the separation of newly formed chromosomes. As in the case of radiation therapy, normal cells have a higher ability to recoverfrom this damage than cancer cells. For this reason chemotherapy is often used at the maximal tolerated dose, with the consequent side effects on tissues that depend on rapid cell turnover such as the oral and gastrointestinal mucosa, the hair, skin, and nails.
One important aim of current research is to develop drugs that target cancer cells based on the specific dependency of these cells on the oncogenic mutations that they contain (Sawyers 2004) (Figure 4). Such “targeted therapies” are no different from many drugs that are available against other types of diseases. Targeted drugs against cancer aim at achieving higher therapeutic effectiveness with fewer side effects. In combination with these drugs, conventional chemotherapy may be applied at lower levels, also with fewer side effects. Targeted therapies include chemical compounds that generally act by inhibiting the enzymatic activity of oncogene products, and monoclonal antibodies that act by blocking oncogenic receptor on the surface of the cell or by antibody-mediated killing of the destruction of the target cell.
The advent of targeted therapies started in the 1990s as a direct result of the identification of critical cancer genes. The new ability to molecularly analyze tumors is revolutionizing tumor classification, prognosis, and treatment. It remains an area of intense research and high promise. Among the monoclonal antibodies, the anti-HER2 antibody trastuzumab (Herceptin™) is effective against a subtype of breast carcinomas that contain an activated HER2 oncogene (Hudis 2007; Shawver et al. 2002). The anti-CD20 antibody rituximab (Rituxan™) is used to treat B-cell lymphomas that present the CD20 antigen (Cheson and Leonard 2008), and the anti-EGFR antibody cetuximab (Erbitux™) is used against advanced colon cancer (Mendelsohn and Baselga 2006) (Figure 4).
Among the targeted chemical compounds, imatinib (Gleevec™), which is a small-molecule inhibitor of the oncogenic BCR-ABL kinase, is successfully used against leukemias that are caused by this oncogene (Schiffer 2007). The EGFR inhibitor erlotinib (Tarceva™) is used against lung carcinomas that are driven by a mutant EGFR (Ciardiello and Tortora 2008). Moreover, although different cancer subtypes in a given organ may have very different sets of driving mutations, certain cancer subtypes in different organs may surprisingly share common mutations. As a result, the same drug may be effective on molecularly related tumors in different organs. A related class of anti-cancer compounds is the angiogenesis inhibitors, which prevent the formation of blood capillaries that feed tumors. Some, such as the monoclonal antibody bevacizumab (Avastin™), are in clinical use (Ferrara 2002) (Figure 4). However, this drug has met with limited clinical success because cancer cells have multiple ways to stimulate angiogenesis (Meyerhardt and Mayer 2005). Current investigation is focusing on identifying combinations of angiogenic inhibitors that would be effective (Bergers and Hanahan 2008).
The barriers to improving the treatment of cancer still remain difficult, which underscores the need to add new directions to the future of cancer therapy. What changes in oncology can be envisioned? In the not too distant future, a patient’s tumor profile could include not only histopathological grading and information on the status of common oncogenic mutations but also a full molecular portrait of the tumor (Massagué 2007; Nevins and Potti 2007). Recent progress in molecular profiling of tumors has led to the discovery of gene-expression signatures that allow a better classification of tumors into distinct subtypes, a better prediction of the risk and site of metastasis, and better identification of relevant therapeutic targets (Bild et al. 2006; Fan et al. 2006; Minn et al. 2005; Padua et al. 2008; van ‘t Veer et al. 2002; van de Vijver et al. 2002). A 70-gene “poor-prognosis” signature (MammaPrint) and a non-overlapping set of 21 “recurrence” genes (Oncotype Dx) have been turned into commercial products that assist clinicians in decisions to spare breast cancer patients with good-prognosis tumors from chemotherapy when this treatment is not required. Genes from these signatures can then be directly tested for their ability to mediate metastasis and to serve as targets of drugs that diminish the metastatic activity of cancer cells (Gupta et al. 2007). Drug regimens for cancer patients might include individualized multi-drug combinations targeting specific disease subtypes and metastatic sites. Betters biomarkers of drug response in patients will help better assess the response of individual patients to targeted therapies (Sawyers 2008).
With the recent new knowledge about the molecular, genetic, and cellular bases for cancer development and progression comes new opportunities to improve and expand our ability to prevent, detect, and treat this disease. Working closely together, clinicians and scientists can generate and apply the necessary knowledge to relegate cancer to the status of one more curable or indolent disease in the next few decades.
Bibliography
American Cancer Society. Cancer facts and figures, 2008. http://www.cancer.org/.
Bergers, G., and D. Hanahan. “Modes of resistance to anti-angiogenic therapy.” Nature Reviews Cancer 8, 2008, 592–603.
Bierie, B., and H. L. Moses. “Tumour microenvironment: TGFbeta: the molecular Jekyll and Hyde of cancer.” Nature Reviews Cancer 6, 2006, 506–520.
Bild, A. H., G. Yao, J. T. Chang, Q. Wang, A. Potti, D. Chass, M. B. JoshI, et al. “Oncogenic pathway signatures in human cancers as a guide to targeted therapies.” Nature 439, 2006, 353–357.
Billingsley, K. G., M. E. Burt, E. Jara, R. J. Ginsberg, J. M. Woodruff, D. H. Leung, and M. F. Brennan. “Pulmonary metastases from soft tissue sarcoma: analysis of patterns of diseases and postmetastasis survival.” Annals of Surgery 229, 1999, 602–610; discussion 610–602.
Blasco, M. A. “The epigenetic regulation of mammalian telomeres.” Nature Reviews Genetics 8, 2007, 299–309.
Boshoff, C., and Weiss, R. “AIDS-related malignancies.” Nature Reviews Cancer 2, 2002, 373–382.
Cano, A., M. A. Pérez–Moreno, I. Rodrigo, A. Locascio, M. J. Blanco, M. G. Del Barrio, F. Portillo, and M. A. Nieto. “The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression.” Nature Cell Biology 2, 2000, 76–83.
Cheson, B. D., and J. P. Leonard. “Monoclonal antibody therapy for B-cell non-Hodgkin’s lymphoma.” The New England Journal of Medicine 359, 2008, 613–626.
Cheung, T. K., H. H. Xia, and B. C. Wong. “Helicobacter pylori eradication for gastric cancer prevention.” Journal of Gastroenterology 42, Suppl 17, 2007, 10–15.
Christofori, G. “New signals from the invasive front.” Nature 441, 2006, 444–450.
Ciardiello, F., and G. TORTORA. “EGFR antagonists in cancer treatment.” The New England Journal of Medicine 358, 2008, 1160–1174.
Clark, E. A., T. R. Golub, E. S. Lander, and R. O. Hynes. “Genomic analysis of metastasis reveals an essential role for RhoC.” Nature 406, 2000, 532–535.
Clarke, M.F., and M. Fuller. “Stem cells and cancer: two faces of eve.” Cell 124, 2006, 1111–1115.
Classon, M., and E. Harlow (2002). “The retinoblastoma tumour suppressor in development and cancer.” Nature Reviews Cancer 2, 2002, 910–917.
Danaei, G., S. Vander Hoorn, A. D. Lopez, C. J. Murray, and M. Ezzati. “Causes of cancer in the world: comparative risk assessment of nine behavioural and environmental risk factors.” Lancet 366, 2005, 1784–1793.
Chapelle, A. de la. “Genetic predisposition to colorectal cancer.” Nature Reviews Cancer 4, 204, 769–780.
Esteller, M. “Cancer epigenomics: DNA methylomes and histone-modification maps.” Nature Reviews Genetics 8, 2007, 286–298.
Fan, C., D. S. Oh, L. Wessels, B. Weigelt, D. S. Nuyten, A. B. Nobel, L. J. Van’t Veer, and C. M. Perou. “Concordance among gene-expression-based predictors for breast cancer.” The New England Journal of Medicine 355, 2006, 560–569.
Ferrara, N. “VEGF and the quest for tumour angiogenesis factors.” Nature Reviews Cancer 2, 2002, 795–803.
Fidler, I. J. “The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited.” Nature Reviews Cancer 3, 2003, 453–458.
Fodde, R., R. Smits, and H. Clevers. “APC, signal transduction and genetic instability in colorectal cancer.” Nature Reviews Cancer 1, 2001, 55–67.
Gavrilovic, I. T., and J. B. Posner, J.B. “Brain metastases: epidemiology and pathophysiology.” J Neurooncology 75, 2005, 5–14.
Gupta, G. P., and J. Massagué. “Cancer metastasis: building a framework.” Cell 127, 2006, 679–695.
—, D. X. Nguyen, A. C. Chiang, P. D. Bos, J. Y. Kim, C. Nadal, R. R. Gomis, K. Manova-Todorova, and J. Massagué. “Mediators of vascular remodelling co-opted for sequential steps in lung metastasis.” Nature 446, 2007, 765–770.
Halazonetis, T.D., V. G. Gorgoulis, and J. Bartek. “An oncogeneinduced DNA damage model for cancer development.” Science 319, 2008, 1352–1355.
Hanahan, D., and R. A. Weinberg. (2000). “The hallmarks of cancer.” Cell 100, 2000, 57–70.
Hausen, H. zur. “Viruses in human cancers.” European Journal of Cancer 35, 1999, 1174–1181.
Hess, K.R., G. R. Varadhachary, S. H. Taylor, W. Wei, M. N. Raber, R. Lenzi, and J. L. Abbruzzese. “Metastatic patterns in adenocarcinoma.” Cancer 106, 2006, 1624–1633.
Hudis, C.A. “Trastuzumab–mechanism of action and use in clinical practice.” The New England Journal of Medicine 357, 2007, 39–51.
Husemann, Y., J. B. Geigl, F. Schubert, P. Musiani, M. Meyer, E. Burghart, G. Forni, et al. “Systemic spread is an early step in breast cancer.” Cancer Cell 13, 2008, 58–68.
Jemal, A., T. Murray, E. Ward, A. Samuels, R. C. Tiwari, A. Ghafoor, E. J. Feuer, and M. J. Thun. “Cancer statistics, 2005.” CA: a cancer journal for clinicians 55, 2005, 10–30.
Joyce, J. A. “Therapeutic targeting of the tumor microenvironment.” Cancer Cell 7, 2005, 513–520.
Kaelin, W. G. “The von Hippel-Lindau tumor suppressor protein: roles in cancer and oxygen sensing.” Cold Spring Harbor Symposia in Quantitative Biology 70, 2005, 159–166.
Kang, Y., P. M. Siegel, W. Shu, M. Drobnjak, S. M. Kakonen, C. Cordon-Cardo, T. A. Guise, and J. Massagué. “A multigenic program mediating breast cancer metastasis to bone.” Cancer Cell 3, 2003, 537–549.
Karin, M. “Nuclear factor-kappaB in cancer development and progression.” Nature 441, 2006, 431–436.
Karpozilos, A., and N. Pavlidis. “The treatment of cancer in Greek antiquity.” European Journal of Cancer 40, 2004, 2033–2040.
Kim, M., J. D. Gans, C. Nogueira, A. Wang, J. H. Paik, B. Feng, C. Brennan, et al. “Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene.” Cell 125, 2006, 1269–1281.
Klein, C. A., T. J. Blankenstein, O. Schmidt-Kittler, M. Petronio, B. Polzer, N. H. Stoecklein, and G. Riethmuller. “Genetic heterogeneity of single disseminated tumour cells in minimal residual cancer.” Lancet 360, 2002, 683–689.
Leiter, U., F. Meier, B. Schittek, and C. Garbe. “The natural course of cutaneous melanoma.” Journal of Surgical Oncology 86, 2004, 172–178.
Lewis, C. E., and J. W. Pollard. “Distinct role of macrophages in different tumor microenvironments.” Cancer Research 66, 2006, 605–612.
Malumbres, M., and M. Barbacid. “Cell cycle kinases in cancer.” Current Opinion in Genetics and Development 17, 2007, 60–65.
Martin, R. W., P. P. Connell, and D. K. Bishop. “The Yin and Yang of treating BRCA-deficient tumors.” Cell 132, 2008, 919–920.Massagué, J. “G1 cell-cycle control and cancer.” Nature 432, 2004, 298–306.
—, “Sorting out breast-cancer gene signatures.” The New England Journal of Medicine 356, 2007, 294–297.
—, “TGFbeta in Cancer.” Cell 134, 2008, 215–230.
Melo, J. V., and D. J. Barnes. “Chronic myeloid leukaemia as a model of disease evolution in human cancer.” Nature Reviews Cancer 7, 2007, 441–453.
Mendelsohn, J., and J. Baselga. “Epidermal growth factor receptor targeting in cancer.” Seminars in Oncology 33, 2006, 369–385.
Meyerhardt, J. A., and R. J. Mayer. “Systemic therapy for colorectal cancer.” The New England Journal of Medicine 352, 2005, 476–487.
Minn, A. J., G. P. Gupta, D. Padua, P. D. Bos, D. X. Nguyen, D. Nuyten, B. Kreike, et al. “Lung metastasis genes couple breast tumor size and metastatic spread.” Proceedings of the National Academy of Sciences USA 104, 2007, 6740–6745.
Minn, A. J., G. P. Gupta, P. M. Siegel, P. D. Bos, W. Shu, D. D. Giri, A. Viale, A. B. Olshen, W. L. Gerald, and J. MASSAGUÉ. “Genes that mediate breast cancer metastasis to lung.” Nature 436, 2005, 518–524.
Mueller, M. M., and N. E. Fusenig. Friends or foes—bipolar effects of the tumour stroma in cancer. Nature Reviews Cancer 4, 2004, 839–849.
Nevins, J. R., and A. Potti. “Mining gene expression profiles: expression signatures as cancer phenotypes.” Nature Reviews Genetics 8, 2007, 601–609.
Norton, L., and J. Massagué “Is cancer a disease of self-seeding?” Nature Medicine 12, 2006, 875–878.
Nowell, P.C. “The clonal evolution of tumor cell populations.” Science 194, 1976, 23–28.
Olsson, A.K., A. Dimberg, J. Kreuger, and L. Claesson-Welsh. “VEGF receptor signalling—in control of vascular function.” Nature Reviews Molecular Cell Biology 7, 2006, 359–371.
Padua, D., X. H. Zhang, Q. Wang, C. Nadal, W. L. Gerald, R. R. Gomis, and J. Massagué. “TGFbeta primes breast tumors for lung metastasis seeding through angiopoietin-like 4.” Cell 133, 2008, 66–77.
Paget, S. “The distribution of secondary growths in cancer of the breast.” Lancet 1, 1889, 571–573.
Parato, K. A., D. Senger, P. A. Forsyth, and J. C. Bell. “Recent progress in the battle between oncolytic viruses and tumours.” Nature Reviews Cancer 5, 2005, 965–976.
Perl, A. K., P. Wilgenbus, U. Dahl, H. Semb, and G. Christofori. “A causal role for E-cadherin in the transition from adenoma to carcinoma.” Nature 392, 1998, 190–193.
Pouyssegur, J., F. Dayan, and N. M. Mazure. “Hypoxia signalling in cancer and approaches to enforce tumour regression.” Nature 441, 2006, 437–443.
Roden, R., A. Monie, and T. C. WU. “The impact of preventive HPV vaccination.” Discovery Medicine 6, 2006, 175–181.
Rustgi, A. K. “The genetics of hereditary colon cancer.” Genes and Development 21, 2007, 2525–2538.
Sawyers, C. “Targeted cancer therapy.” Nature 432, 2004, 294–297.
Sawyers, C. L. “The cancer biomarker problem.” Nature 452, 2008, 548–552.
Schiffer, C. A. “BCR–ABL tyrosine kinase inhibitors for chronic myelogenous leukemia.” The New England Journal of Medicine 357, 2007, 258–265.
Schmidt–Kittler, O., T. Ragg, A. Daskalakis, M. Granzow, A. Ahr, T. J. Blankenstein, M. Kaufmann, et al. “From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression.” Proceedings of the National Academy of Sciences USA 100, 2003, 7737–7742.
Shawver, L.K., D. Slamon, and A. Ullrich. “Smart drugs: tyrosine kinase inhibitors in cancer therapy.” Cancer Cell 1, 2002, 117–123.
Sweet-Cordero, A., S. Mukherjee, A. Subramanian, H. You, J. J. Roix, C. Ladd–Acosta, J. Mesirov, T. R. Golub, and T. Jacks. “An oncogenic KRAS2 expression signature identified by cross-species gene-expression analysis.” Nature Genetics 37, 2005, 48–55.
Thiery, J.P. “Epithelial-mesenchymal transitions in tumour progression.” Nature Reviews Cancer 2, 2002, 442–454.
Veer, L. J. van’t, H. Dai, M. J. van de Vijver, Y. D. He, A. A. Hart, M. Mao, H. L. Peterse, K. van der Kooy, M. J. Marton, A.T. Witteveen, et al. “Gene expression profiling predicts clinical outcome of breast cancer.” Nature 415, 2002, 530–536.
Vijver, M. J. van de, Y. D. He, L. J. van’t Veer, H. Dai, A. A. Hart, D. W. Voskuil, G. J. Schreiber, et al. “A gene-expression signature as a predictor of survival in breast cancer.” The New England Journal of Medicine 347, 2002, 1999–2009.
Vogelstein, B., E. R. Fearon, S. R. Hamilton, S. E. Kern, A. C. Preisinger, M. Leppert, Y. Nakamura, R. White, A. M. Smits, and J. L. Bos. “Genetic alterations during colorectal-tumor development.” The New England Journal of Medicine 319, 1988, 525–532.
—, K. W. Kinzler. “Cancer genes and the pathways they control.” Nature Medicine 10, 2004, 789–799.
Vousden, K.H., and D. P. Lane. “p53 in health and disease.” Nature Reviews Molecular Cell Biology 8, 2007, 275–283.
Walsh, T., and M. C. King. “Ten genes for inherited breast cancer.” Cancer Cell 11, 2007, 103–105.
Wang, C., Y. Yuan, Y., and R. H. Hunt. “The association between Helicobacter pylori infection and early gastric cancer: a meta-analysis.” The American Journal of Gastroenterology 102, 2007, 1789–1798.
Wang, J., R. Loberg, and R. S. Taichman, R.S. “The pivotal role of CXCL12 (SDF–1)/CXCR4 axis in bone metastasis.” Cancer Metastasis Reviews 25, 2006, 573–587.
Wang, W. “Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins.” Nature Reviews Genetics 8, 2007, 735–748.
Weinberg, R. A. The biology of cancer. New York: Garland Science, 2007.
Welcsh, P. L., and M. C. King. “BRCA1 and BRCA2 and the genetics of breast and ovarian cancer.” Human Molecular Genetics 10, 2001, 705–713.
Woodman, C. B., S. I. Collins, and L. S. Young, L.S. “The natural history of cervical HPV infection: unresolved issues.” Nature Reviews Cancer 7, 2007, 11–22.
World Heath Organization. Cancer. Fact Sheet No 297, 2008. http://www.who.int/mediacentre/factsheets/fs297/en/index.html.
Yang, J., S. A. Mani, J. L. Donaher, S. Ramaswamy, R. A. Itzykson, C. Come, P. Savagner, I. Gitelman, A. Richardson, and R. A. Weinberg. “Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis.” Cell 117, 2004, 927–939.
Young, L.S., and A. B. Rickinson. “Epstein-Barr virus: 40 years on.” Nature Reviews Cancer 4, 2004, 757–768.
Comments on this publication