Since the announcement of Dolly in 1997, cloning by nuclear transfer has received considerable media attention. But commentators have often been preoccupied with speculations about armies of cloned dictators or sportsmen, and minor applications such as replacement pets. Here we wish to place nuclear transfer in a broader context and outline the more subtle, but profound consequences of the work.
So, why was such an elaborate way of producing animals actually developed? Visitors to Scotland will notice there is hardly a shortage of sheep. There were in fact two motivations behind the Dolly experiment. One was strictly commercial, to develop a tool for rapid production of identical animals for biotechnology. The second and more powerful impulse was basic scientific curiosity and an opportunity to address a long-standing biological question. As complex animals, frogs, mice, sheep, and people arise from a single cell, and many different cell types are formed; how do they adopt their different fates and how do they maintain or change their identity?
Early investigations and founding principles
Scholars have pondered the question of animal development since ancient times. In the third century BC, Aristotle recognized the importance of sexual reproduction and proposed two alternative models. Either the structure of the whole animal is already preformed in miniature within the egg or embryo, or new structures arise progressively. Aristotle favored the second idea, but without suitable technology the question remained the subject of philosophical debate for centuries. Preformationism became the favored view in seventeenth- and eighteenth-century Europe, as illustrated by the seventeenth-century engraving in Figure 1. Stimulated by the discovery of sperm, or as they were termed at the time “animalcules” in seminal fluid, the physicist and early microscopist Nicholas Hartsoeker speculated about the possible structure of a tiny fetus within. Hartsoeker conjectured that the head of the sperm grew to form the fetus and the tail of the sperm formed the umbilical chord, while the function of the egg was merely to provide a nest supporting its development.
Reliable observations however only became possible after 1830 with the invention of the compound microscope by the British amateur naturalist Joseph Jackson Lister. Multiple lens instruments provided sufficient resolution to make out the detailed structure of living material for the first time. Modern biology arguably began in 1839 when Theodor Schwann and Matthias Schleiden demonstrated that living things are composed of cells. Shortly after, Albrecht von Kölliker showed that sperm and eggs (oocytes) are also cells, but how they interact to form a new organism was a mystery. The eminent chemist Justus von Liebig proposed that sperm transmit their male qualities to the oocyte through the vigorous vibrations of their tails. Then in 1854, George Newport described his observations of fertilization in frogs and suggested that sperm make their contribution by actually penetrating the egg. Around the same time, microscopic investigations were revealing that new cells arose by division of the fertilized egg, making it unlikely that development occurs by preformation.
Oskar Hertwig is credited with beginning the study of fertilization and early embryo development in the sea urchin, a very productive field that provided much of the information subsequently applied to other species. Sea urchins are ideal for microscopy because the eggs are very clear. In 1876, Hertwig described his observations of events following the addition of sperm to eggs. In particular, he noted the presence of two nuclei in the egg, one of which came from the sperm, and reported how they fused together. This provided the first explanation for the role of the two parents. It also focused attention on the importance of the nucleus, and the colored bodies within that could be revealed using the newly developed aniline dyes, and named “chromosomes” in the 1880s.
The German biologist August Weismann ranks perhaps second only to Charles Darwin in his contributions to theoretical biology. In 1892 Weismann made the bold proposal that the nuclei of eggs and sperm contain a hereditary substance, and that this constitutes the only organic continuity between generations (Weismann 1892). This principle laid the foundations for all of genetics and evolutionary biology. Weismann’s “germ-plasm theory” states that the germ cells are a lineage quite distinct from the other cells of the body, the somatic cells, and that characteristics acquired by the body during life are not transmitted to the germ cells. This was an explicit rejection of the views of Jean-Baptiste Lamarck that were widely held at the time, including by Darwin himself. Over the next twenty years, the strand of thought commenced by Weismann developed into the modern sciences of genetics and development. In 1900, Gregor Mendel’s work on pea hybrids was rediscovered and with it his concept of discrete segregating traits. Within two years Theodor Boveri and Walter Sutton had both proposed that elements specifying Mendelian traits are located in the chromosomes. In 1907, Boveri demonstrated that a normal set of chromosomes is necessary to support embryonic development in the sea urchin. In 1915, Thomas Morgan described the physical location of genes on the chromosomes of the fruit fly in his masterwork The Mechanism of Mendelian Heredity (Morgan et al. 1915).
These ideas are now the stuff of basic biology courses, but during the twentieth century they were seen by some as “degenerate and fascist.” Germ-plasm theory and all that followed were violently rejected by the ideologues of Soviet Russia. From the early 1930s until as late as 1964, official Soviet policy denied Weismann’s ideas and the whole of genetics. Stalin is not primarily remembered for his interest in developmental biology, and it seems likely that this was just a political convenience. The inheritance of acquired characteristics allowed that the human race could be perfected through “progressive materialist” politics. Soviet leaders could therefore justify the hardships endured by ordinary people as worthwhile and necessary for the production of future generations of ideal Communists. This political atmosphere also favored the rise of the notorious Soviet agronomist Trofim Lysenko. In a ranting address to the Lenin All-Union Academy of Agricultural Sciences in August 1948, Lysenko denounced Weismann at length and mocked the “bourgeois pseudoscience” of his followers. In his ridicule, Lysenko inadvertently provided a reasonable description of the modern concept of the genome and a substance that would become known as DNA:
Weismann denied the inheritability of acquired characters and elaborated the idea of a special hereditary substance to be sought for in the nucleus. The sought for bearer of heredity, he stated, is contained in the chromosome material. […] An immortal hereditary substance, independent of the qualitative features attending the development of the living body, directing the mortal body, […] that is Weismann’s frankly idealistic, essentially mystical conception (Lysenko 1948).
Nature is however brutally indifferent to political theory. Lysenko’s methods were responsible for repeated crop failures in the USSR and, when similar agricultural policies were adopted in China in 1958 under the “The Great Leap Forward,” they contributed to the greatest famine in recorded history, between 1959–61.
Cloning and cell determination
Tied in with his concept of the germ plasm, Weismann offered the first testable theory of animal development, a process termed mosaic development. He proposed that the single cell embryo, the zygote, contains factors or determinants localized in discrete regions. As it cleaves, the determinants are distributed unequally between daughter cells and control their future development. The process continues as the various cell types form by “differentiation”, as the embryo develops. This model clearly predicts that individual cells of the developing embryo should not share the same potential. However in 1892, Hans Driesch provided the first evidence against Weismannís theory (Driesch 1892). Cells of early sea urchin embryos could be separated and each form a whole larva. Division at the two-cell stage led to two normal larvae and individual cells from the four-cell stage produced four normal larvae. These were in fact the first experimentally cloned animals.
In a lecture presented at London University in October 1913, Driesch stated that the embryo is a “harmonious equipotential system […] each element of which is capable of playing a number of different roles. The actual role it plays in any given case being a function of its position.” Since then, there have been many demonstrations that the embryos of many vertebrates, including mammals, can be reorganized by changing the arrangement or the number of cells, and then recover to form a normal whole animal.
Cloning by nuclear transfer was first proposed as a further means of testing whether nuclei from early and late embryonic cells had equivalent developmental potential, and is a rather older idea than often supposed. Yves Delage, a now obscure French marine biologist, made the first recorded reference to the procedure in 1895, claiming “if, without any deterioration, the egg nucleus could be replaced by the nucleus of an ordinary embryonic cell, we should probably see this egg developing without changes.” (Beetschen and Fischer 2004) However Delage is not known to have carried out such an experiment. The honor usually goes to Hans Spemann, a former student of Boveri. In 1928 Spemann performed the first nuclear transfer with a remarkable piece of microsurgery (Spemann 1928). Spemann’s own drawings are shown in Figure 2. Using micro-tweezers and a loop of hair from his baby daughter, he constricted a single cell salamander embryo into two parts, one of which contained the cell nucleus (Figure 2CA). Left to develop, the portion with the nucleus divided and formed an embryo, while the other side remained a pouch of clear cytoplasm (Figures 2CB and 2C). The embryo was allowed to develop to later stages, such as 16-cell, at which time a single nucleus was allowed to pass back into the empty cytoplasm (Figure 2D). The single cell then developed into a normal salamander embryo at a slightly earlier stage, (Figure 2E). This clearly proved that later embryonic cell nuclei are capable of forming a complete animal.
It is not known whether Spemann knew of Delage’s earlier proposal, but in 1936 after his retirement, Spemann proposed what he termed “a fantastical cloning experiment.” (Spemann 1936) If one could transfer nuclei from cells at yet later stages of development back into fertilized eggs it would be possible to systematically track when cells retained or lost their ability to form a whole organism, a quality now termed “totipotency.”
The next decades continued to see many developmental biologists focus on sea urchins and amphibians, because they were relatively easy to culture and manipulate. Frog oocytes are very large cells, around 1–2 millimeters in diameter, and quite prominent as dark grey or brown spheres within their protective jelly coat. In the early 1950s, Robert Briggs and Thomas King carried out Spemann’s fantastical experiment with frogs (Briggs and King 1952). They removed the nucleus from an activated oocyte, using a glass needle. A single cell dissected from a later stage embryo was then drawn up into a fine glass pipette connected by rubber tubing to an ordinary syringe. The cell broke open as it was squeezed within the pipette and the free nucleus was injected into the enucleated egg. Culturing the reconstructed embryos further, they found that cell nuclei from blastula stage embryos could direct normal development to feeding-stage larvae. But nuclei from later stage embryos, in which the major embryonic cell lineages such as mesoderm or endoderm were already established, were unable to do so.
John Gurdon and Ron Laskey later extended the work using nuclei from juvenile and adult tissues, such as the skin of the foot web, and generally found that these animals survived up to tadpole stage, but not much further. Gurdon did derive some adult frogs from tadpole intestinal tissue in 1962 (Gurdon 1962), but the possibility that germ cells were present in his tissue left the results in doubt. The overwhelming view at the time was that the developmental capacity of transplanted nuclei decreased with the age and extent of differentiation of the donor cell. Nuclei of the very early embryo may be equivalent, but at some stage their fate becomes determined, “hard wired” by some concrete change, such as the loss or irreversible modification of DNA in the nucleus.
This view was however difficult to reconcile with some well-known phenomena, notably the regenerative capabilities of most fish, and amphibians such as newts and salamanders. If a newt loses a limb, cells from surrounding tissues such as the skin migrate into the wound and undergo a process of “reverse development” dedifferentiating to form a blastema. This is a mass of undifferentiated cells that divide rapidly. Cells within the blastema then differentiate and re-organise to form a replacement limb. This was good evidence that some adult differentiated cells are not determined in their fate and can radically change their identity. Was limb regeneration fundamentally different to generating a whole animal by nuclear transfer? Or was the failure of nuclear transfer a result of technical rather than biological limitations? These open questions provided sufficient motivation for some researchers to continue probing cell determination.
Sheep lead the way
Most biologists are mammals, and naturally keen to investigate species closer to themselves than sea urchins and amphibians, but for many years this was just too difficult technically. Mammalian embryos grow within the controlled conditions of the female reproductive tract rather than pond or seawater and, although large compared to other cells at about one-tenth of a millimeter across, are barely visible to the naked eye. It took until the 1970s and 80s, when embryo culture and micromanipulation techniques had improved sufficiently, for transfer of mammalian nuclei to become practical. The basic idea remained as Spemann had conceived it, the genetic material is removed from an egg, and then replaced with the nucleus of another cell, often by fusing the whole cell with the oocyte.
The natural focus was on the favorite laboratory mammal, the mouse. However, attempts to repeat Briggs and Kings work in mice were repeatedly unsuccessful. In 1981, Karl Illmensee and Peter Hoppe claimed that they had cloned mice by transfer of nuclei from blastocyst stage embryos (Illmensee and Hoppe 1981). However, their work was later investigated and determined as false, although deliberate fraud was never proven. Then in 1984, James McGrath and Davor Solter seemed to put an end to mammalian nuclear transfer. They systematically transferred nuclei from 1-, 2-, 4-, 8-cell and blastocyst stage embryos into enucleated zygotes, 1-cell stage embryos. Nuclei from 1-cell embryos supported development to blastocysts, success dropped off sharply using 2-cell stage nuclei and failed entirely with later stages. This they reasonably interpreted as a rapid loss of totipotency during development. Their paper concludes with the categorical statement that “the cloning of mammals by simple nuclear transfer is biologically impossible.” (McGrath and Solter 1984)
In retrospect, it was unfortunate that so many early efforts focused on mice. It has since become clear that they are one of the more difficult species to clone by nuclear transfer. This meant that, somewhat unusually, major breakthroughs were made using livestock. The first nuclear transfer mammals were three Suffolk sheep produced by Steen Willadsen, by merging single cells from 8-cell embryos with enucleated unfertilised eggs (Willadsen 1986). Ironically these lambs were born in 1984, just a few months before McGrath and Solter declared mammalian cloning impossible. The reason for this discrepancy was technical. McGrath and Solter had used enucleated zygotes for nuclear transfer, because mouse oocytes are too fragile to survive nuclear transfer. Willadsen had been able to use unfertilized oocytes, which are more robust in sheep. Years of work have since shown that unfertilized oocytes are successful recipients for nuclear transfer in numerous species, while zygotes can only be used at a very particular stage. Only this year has a model been proposed to explain this difference, as we discuss later (Egli, Birkhoff, and Eggan 2008).
During the next decade nuclear transfer was carried out in a variety of mammals, but, as in the frog, it was only successful using cells obtained directly from very early embryos, or cultured for very short periods.
In the early nineties, Keith Campbell and Ian Wilmut of the Roslin Institute near Edinburgh began to study how the choice of oocyte recipient and the cell cycle stage of the nuclear donor cell affected the outcome of nuclear transfer. This made a very significant contribution to the eventual success of nuclear transfer in mammals, so here we outline the main points.
Figure 3. Nuclear transfer and the cell cycle. A) Normal fertilization and development to the 2-cell stage embryo. B) The cell cycle. G1 phase is followed by S phase where the cell duplicates each chromosome, then G2 phase, and mitosis (M) in which the nucleus breaks down, the duplicated chromosomes condense, align on the spindle and are distributed into two new daughter cells. C) Nuclear transfer using a donor cell in G1 phase and development to 2-cell stage embryo. 1n, 2n, 4n = copies of each chromosome.
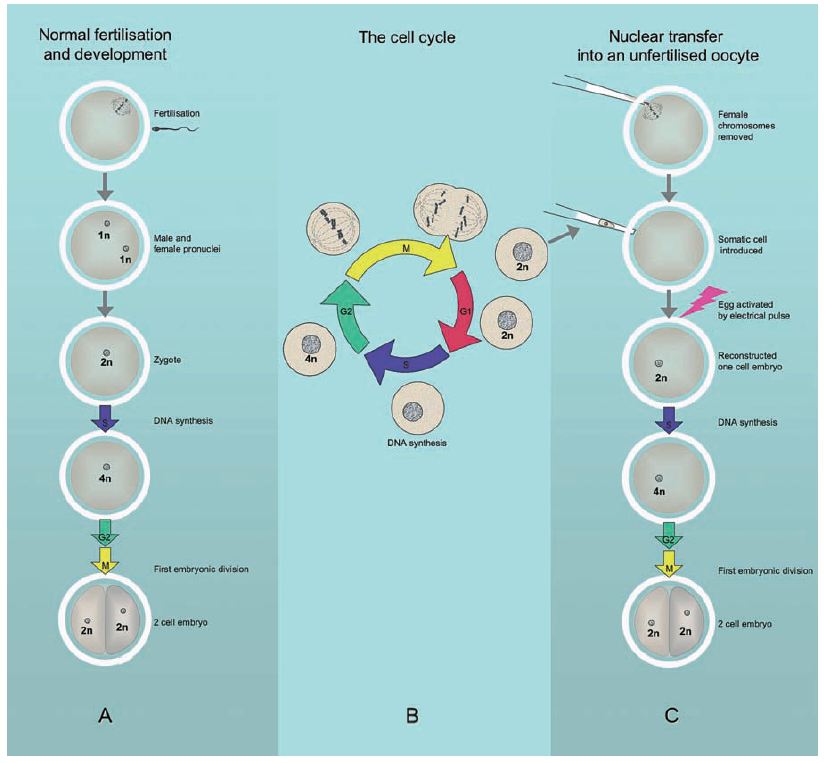
Mammalian oocytes are formed from the germ cells by a process termed meiosis, which leaves each oocyte with only one copy of each chromosome, usually abbreviated as “1n.” When the head of the sperm enters, it causes the oocyte to complete meiosis and initiates the first events of development, illustrated in Figure 3A. The two clusters of male and female chromosomes first form into separate pro-nuclei, which then join to create a single nucleus, in what is now the one cell embryo or zygote. All chromosomes are then replicated by DNA synthesis ready for the first embryonic cell division.
This first division and all subsequent cell divisions that form and maintain the body of the animal is by a process termed mitosis. Mitosis is part of a cycle that ensures that dividing cells maintain the correct number of chromosomes. This “cell cycle” is conventionally divided into four phases, as outlined in Figure 3B. The first phase is termed gap1 (G1), during which the cell has two copies of each chromosome (2n). In the next phase, synthesis (S), the cell replicates all its DNA. Then follows gap2 (G2) phase, when each chromosome is present as four copies (4n). At mitosis (M), the nucleus breaks down, the duplicated chromosomes condense, align on a structure termed the spindle, and are then pulled apart into two new daughter cells. Each new cell contains 2n chromosomes and the process repeats. In rapidly dividing cells the whole cycle takes about one day.
This has profound implications for nuclear transfer. When a cell nucleus is transferred into an unfertilized oocyte it has to be artificially activated, e.g. by an electrical pulse, to kick start development. This initiates DNA synthesis in readiness for the first cell division. However, this occurs regardless of the cell cycle stage of the donor nucleus. If the incoming nucleus was in S or G2 phase, when DNA has already been partially or completely replicated, its DNA will be re-replicated, leading to incorrectly high chromosome numbers, or serious chromosomal damage. The key insight by Campbell and Wilmut was that only donor cells in G1 phase (prior to DNA replication) would support normal development in unfertilized oocytes, as shown in Figure 3C.
The method they developed, and still widely used, is to starve the donor cells of growth factors by reducing the amount of serum in the culture medium for a few days. This blocks the cell cycle before S phase, exactly what is required. Importantly, effective serum starvation requires that cells are cultured for several days.
1995 saw the birth of Megan and Morag at the Roslin Institute, two lambs made by transfer of nuclei from cells grown by Jim McWhir from a day 9 sheep embryo and cultured for 6 to 13 passages (Campbell et al. 1996). These sheep prompted debate amongst the coauthors about what the key factor was that had led to success. Campbell and Wilmut’s view was that serum starvation before nuclear transfer not only coordinated the cell cycle, but also induced a quiescent state in the nucleus making it particularly amenable to reprogramming by the oocyte. McWhir contended that the key lay in some special property of the cells he had derived.
Sheep nuclear transfer is dictated by the natural breeding season and so the question could not be resolved until the next year. The original plan for 1996 was to use embryonic cells again and also test whether the technique could be extended to cells at a later developmental stage, fibroblasts from a day 26 fetus. At that time, we were working with PPL Therapeutics, a biotechnology company in the business of producing pharmaceutical proteins in the milk of transgenic sheep, a short walk away from the Roslin Institute. In a discussion over lunch we suggested a bolder experiment and proposed including adult cells in the 1996 nuclear transfer season. This met with skepticism and the opinion that it was too soon, and anyway no finance was available to extend the experiment. There was however the possibility that if the extra work could be justified in commercial terms, our company might provide funds. As it happened, we were then investigating introducing milk transgenes into sheep mammary epithelial cells derived by Colin Wilde of the Hannah Research Institute in Ayr, as a means of testing their expression. Combining the two projects offered an ideal opportunity. If the mammary cells could be converted into live animals, PPL would have a potential means of producing “instant flocks” of sheep known to express a particular transgene. And, most excitingly, using adult cells for nuclear transfer would address the long-standing question of cell determination. The case was made to the managing and research directors of PPL, Ron James and Alan Colman, and the company took the risk of releasing funds for the experiment. In February 1996, cultures of sheep mammary cells and also cultured embryonic cells were serum-starved and transported over to the Roslin Institute. Bill Ritchie, Ian Wilmut’s skilled technician, then transferred them into Scottish Blackface enucleated oocytes.
A single female lamb was born on July 5, 1996, and named Dolly by the animal caretaker John Bracken, in honor of Dolly Parton and her great singing talent. Two sheep were also born from fetal fibroblasts and four from embryonic cells. This clearly showed that success was clearly not due to any special cell type; the idea that quiescence played a role was also later discarded. What did emerge was the importance of cell synchronization.
A description of the experiment was published on February 27, 1997 (Wilmut et al 1997). More than a decade later, one can calmly state that nuclear transfer from an adult cell caused the concept of irreversible cell determination to be discarded. However, this would be to overlook the sometimes heated contention that raged for 17 months following publication. So fixed was the view of cell determination that several prominent scientists in the US and Europe dismissed the paper outright as a fraud, perhaps recalling the Illmensee controversy. An article in the New York Times from July 29, 1997, gives a sense of the mood: “How do we know the whole thing wasn’t a hoax? Why, some ask, is the rest of the world so willing to accept the world-shattering claim that an adult animal was cloned?”
Other commentators interpreted the inefficiency of adult nuclear transfer as an indication that Dolly was a one-off, an experimental aberration. Several eminent scientists suggested that she was not actually cloned from an adult cell, but had instead arisen from some contaminating embryonic or fetal material. One proposition was that fetal cells present in the blood circulation of the sheep used to provide the mammary cells had somehow carried through into the mammary cell cultures. It seemed that any alternative explanation, no matter how unlikely, was preferable to overturning the doctrine of cell determination. Time magazine ran an article on March 2, 1998, headed “Was Dolly a mistake?” that concluded: “Dolly, in other words, may turn out to be a fluke, not a fake. No matter what she is, it’s looking less and less likely that we’re going to see clones of Bill Gates or Michael Jordan anytime soon.”
Meanwhile, we—along with others—had reported more cloned animals (Schnieke et al. 1997), but these were from cultured fetal cells and so did not confirm adult cloning.
The accusations and speculations thankfully ceased on July 23, 1998. That day’s edition of the journal Nature contained two relevant articles. One gave the results of an independent DNA fingerprinting analysis, confirming that Dolly’s nuclear DNA was identical to the cultured mammary cells (Signer et al. 1998). The second was a report by Ryuzo Yanagimachi and Teruhiko Wakayama of the University of Hawaii describing another animal cloned from adult cells, a mouse named “Cumulina” after the cumulus (ovarian follicle) cells used as donors (Wakayama et al. 1998). The reality of adult cloning was finally accepted. More than a decade has now past and it is timely to revue the developments that followed.
Reproductive cloning
Inevitably, most public, political, and ethical discussions have centered on human reproductive cloning and as a result new laws and regulations are in place around the world. It is perhaps worth emphasizing that, despite announcements from odd cult groups and publicity seekers, none of the scientists actually working in the field ever considered carrying out human reproductive clonin.
As is often the case, once a method is well established, it is difficult to see why it was once viewed as impossible. A variety of different cell types, both fetal and adult, have now been successfully used as nuclear donors and over 20 species cloned, including fish, frogs, fruit flies, and the mammals listed in the table. The efficiency is however low in most species, with only 1–5% of reconstructed embryos proceeding to birth. Nuclear transfer animals can suffer ill health, but their offspring, such as Dolly’s lamb Bonny, do not.
The majority of nuclear transfer experiments are still carried out on livestock. The improvements made have been incremental rather than dramatic, but have nevertheless resulted in success rates of ~15% in cattle. One important factor has been the advance of oocyte maturation techniques. All livestock oocytes are now obtained from ovaries collected from slaughterhouses rather than flushed from the reproductive tract of live animals. This offers an abundant supply of oocytes and greatly reduces the number of animals required. It also makes commercial nuclear transfer viable despite its inefficiency, especially for the reproduction of elite animals with highly desirable characteristics such as racehorses or prize bulls. In the US, companies such as ViaGen offer livestock cloning as part of their assisted reproduction services. Their website (www.viagen.com) states: “ViaGen enables the owners of cattle, horses and pigs to preserve and multiply their best genetics through gene banking and cloning services, and to protect their brands through genomic services.” Devoted (and wealthy) dog owners might also be interested to know that “by calling the toll-free number 888-8VIAGEN you can discuss options for cloning your pet dog.”
Nuclear transfer has been used where normal sexual reproduction is impossible as a result of accident, disease, or natural infertility, as demonstrated by the cloning of a mule (Woods et al. 2003). It has also been applied to reproduce endangered species such as the European wildcat or rare cattle breeds. However, only those species with domesticated relatives available to provide suitable oocytes are likely to benefit. Cloning also cannot improve the genetic diversity of small animal populations, vital for long term survival.
Animals for biomedicine
The future will show whether or not nuclear transfer will become just one more, albeit expensive, routine technique for animal reproduction. But it has already become one of the best methods of founding lines of genetically modified “transgenic” large animals. To produce transgenic animals, there are two choices. Transgene DNA can be introduced directly into the zygote, hoping that it becomes incorporated into the genome. Alternatively, genetic modifications can first be engineered into cultured cells that are then used to produce whole animals. The first method is basically a lottery, animals must be produced before analyzing whether a transgene is present. The second method allows much more control and involves fewer animals, because cells can be thoroughly analyzed in the laboratory before any animals are produced.
In mice, a cell-based method has been available since the early eighties. Mouse embryonic stem (ES) cells can be isolated from early embryos, grown indefinitely in culture, undergo manipulations such as the addition of a transgene or the precise alteration of a particular gene (gene targeting), and then be incorporated back into a developing embryo. The phenomenal power of gene targeting technology in ES cells has provided most of our knowledge about the function of genes in whole animals. This was recognized by the 2007 Nobel Prize for Medicine, awarded jointly to Mario Capecchi, Martin Evans, and Oliver Smithies. It has long been clear to many researchers that extending this to large animals would have many useful applications. But despite considerable efforts, functional ES cells had (and still have) not been derived from livestock. Livestock nuclear transfer using ordinary somatic cells that could be grown and manipulated in culture neatly sidestepped this.
In the experiments directly following Dolly, we demonstrated that both transgenic and gene targeted sheep can be generated by nuclear transfer (Schnieke et al. 1997a; Schnieke et al. 1997b). Since then, many others have followed, e.g. gene targeted cattle resistant to mad cow disease (bovine spongiform encephalopathy) (Kuroiwa et al. 2004). Most applications have however been in the area of biomedicine, including rather later than anticipated, transgenic animals producing pharmaceutical proteins in milk. ATryn, an anticoagulant drug used to treat patients with a hereditary deficiency in the blood protein antithrombin, is being produced in transgenic goats from a cloned founder animal, and was launched on the market in November 2007 by GTC biotherapeutics. Nuclear transfer is also being used to engineer multiple genetic modifications into pigs to provide cells or organs for transplantation into humans, termed xenotransplantation.
A number of large animal models of serious human diseases such as cystic fibrosis (Rogers et al. 2008), diabetes, and various cancers are also being developed. These are often extensions of work first carried out in mice, where gene targeting has provided a huge amount of information regarding diseases such as cancers. Many strains of mice with defined genetic defects have been produced and these have been very valuable in understanding mechanisms such as tumor initiation and progression (Frese and Tuveson 2007). Mice are also useful for proof-of-principle studies for novel diagnostic and treatment strategies, as we discuss later. However the major differences in body size, general physiology, anatomy, diet, and lifespan restrict the usefulness of mice closer to the clinic. For example, radiation and thermal therapy cannot easily be scaled down to treat mouse-sized tumors. Nuclear transfer offers the opportunity to extend the range of genetically-defined disease models to other species, such as pigs, which more closely resemble humans in their size, anatomy, and physiology.
Nuclear transfer, embryonic stem cells, and regenerative medicine
As mentioned above, much of the interest in nuclear transfer in the late eighties and early nineties was prompted by the possibilities offered by ES cell technology. Since then, the two fields have been closely intertwined.
ES cells are usually isolated from blastocyst stage embryos. A blastocyst is a tiny fluid filled ball of about one hundred cells containing within it a clump of cells termed the inner cell mass (ICM) that gives rise to all tissues of the body. Blastocysts, or isolated ICMs, are placed in culture and over a period of several days or a week, colonies of tightly packed small cells emerge and continue to grow indefinitely; these are ES cells. For reasons that are unclear, deriving ES cells is however difficult in many species and has only been achieved in mouse, human, and rhesus monkey. Rat ES cells have been announced to the press, but not yet published in a scientific journal.
ES cells are often used as a convenient surrogate for the study of the early embryo. But what they actually are is still not clear. They may be an artifact of tissue culture, something aberrant created in response to artificial growth conditions. Recent evidence however suggests they are a cell type normally present for a short time in the embryo, which can be captured and maintained by the proper culture conditions (Silva and Smith 2008).
The defining characteristic of ES cells is that they can grow indefinitely as undifferentiated cells and then differentiate to many other cell types. When introduced into a blastocyst they can integrate into the ICM and participate in forming all tissues of the body. When given appropriate stimuli they can also form a wide variety of cell types in culture, so called in vitro differentiation. Since human ES cells were first derived by Jamie Thomson ten years ago (Thomson et al. 1998) there has been intense interest and enthusiasm surrounding in vitro differentiation as a possible source of replacement human tissue, such as nerve cells, insulin producing cells, or heart muscle. Many articles have been written on the subject, so we will not go into detail here. The basic scheme is shown in panel A of Figure 4. The promise of ES based therapy is undoubtedly real, but a few words of caution are perhaps in order. Persuading human ES cells to form useful amounts of an appropriate, properly characterized, and pure therapeutic cell-type remains a very difficult challenge. Rigorous methods also need to be established to ensure that ES derived preparations are free of potentially tumor-forming cells. Research scientists and the biotech industry should be realistic and avoid the tendency to hype their claims.
The Californian pharmaceutical company Geron is perhaps farthest advanced in human ES cell therapies. The company website (www.geron.com) reports the development of human ES derived nerve cell progenitors for acute spinal cord injury and cardiomyocytes for the treatment of heart failure. Although Geron have applied for permission to carry out human clinical trials of their nerve cell progenitors, the United States Food and Drug Administration have placed the application on hold. If and when trials are approved, the outcome will be very important for the future of ES cell therapy.
If they can be produced, ES derived tissues would need to be immunologically matched to the patient in the same way as ordinary donated tissue to avoid rejection. Recipients are also likely to require lifelong immune suppression. Tissue matching is a particular problem for patients with unusual tissue types, such as people of mixed race. Shortly after Thomson’s report, it was suggested that nuclear transfer could provide a means of producing tailor-made human tissues by “therapeutic cloning.” Cells could be taken from a human patient who needed tissue replacement therapy and used to produce cloned embryos. ES cells would be derived and then induced to differentiate in culture. The replacement tissue would be perfectly matched to the patient’s own body, see Figure 4B.
Some of the necessary technical steps have been achieved in animals. For example, ES cells have been derived from nuclear transfer mouse embryos and found to be the same as those from normal embryos. Rhesus monkey ES cells have also been produced from cloned embryos, but so far no human “NT ES” cells have been derived. The major practical problem is the supply of unfertilised human oocytes, which is already insufficient to meet the existing needs of people requesting assisted reproduction techniques such as in vitro fertilisation (IVF). A recent news item in Nature revealed that, despite two years and $100,000 spent on local advertising, stem cell researchers at Harvard University have managed to secure only one egg donor (Maher 2008).
Therapeutic cloning is therefore unlikely to be realized unless an alternative source of recipient oocytes can be found. Animal, particularly cattle, oocytes are plentiful thanks to in vitro maturation. The UK Human Fertilization and Embryology Authority (HFEA) recently approved research into whether these could be used, but many people object to the creation of cytoplasmic hybrid embryos. Biological problems may also arise from incompatibility between the components of the animal oocyte and the incoming human nucleus. Reprogramming factors and important cellular organelles such as the mitochondria may not function properly. Perhaps the most promising source of oocytes is in vitro maturation of immature human oocytes from donated ovaries. Although less advanced than in cattle, in vitro maturation of human oocytes is improving, being used mainly to help women who must undergo ovariectomy. Several normal births from in vitro matured oocytes have been reported.
Despite their possible benefits, human embryonic stem cell derivation and therapeutic cloning both face serious ethical and religious opposition, and it is clear that many people will accept neither the artificial generation nor destruction of human embryos, even if only tiny balls of cells. Laws vary around the world, for example the UK HFEA gave approval in 2007, the Spanish Ministry for Health approved the work in 2008, while Germany has no plans to legalize the procedure. Another problem is the high cost and considerable time required. Therapeutic cloning would most probably be restricted to wealthy patients and then only those whose disease condition allowed them to wait several months. This said, recent advances in nuclear reprogramming have probably already made therapeutic cloning obsolete.
Understanding reprogramming
A human body contains several hundred cell types, each of which differ in a multitude of cellular components. The identity of a cell, its shape, how fast it divides, the materials it synthesizes, the receptors on its surface, and the multifarious nanomachines we call RNA and protein molecules, are all ultimately the product of different patterns of gene expression.
Cloning from adult cells showed that these patterns are not due to immutable genetic differences. The nuclei of even the most highly differentiated cells, such as neurons, or mature B-lymphocytes specialized to synthesize a single antibody, retain the potential to form all the cells of the body (Hochedlinger and Jaenisch 2002). Each cell has the same genetic information, in humans around three billion DNA base pairs and an estimated 25,000 genes, but these are expressed in different ways. A parallel may be drawn with computer software and hardware. Different programs dealing with graphics, mathematics, music, or word processing can all run on the same machine without altering the physical components. Of course, unlike computers, cells are self-organizing and no one would suggest that a cell receives a complete set of instructions from some outside agency.
The regulation of gene expression has been studied for over thirty years, and is known to operate at many levels: from the accessibility of the DNA within the nucleus to expression factors; the rate at which genes are transcribed into messenger RNA molecules; the processing and transport of RNA; to the synthesis and degradation of protein products. A glance through the diagrams in a modern molecular or cell biology textbook reveals multitudes of arrows symbolizing the regulatory pathways and feedback loops that govern the workings of our cells. Terms often used are “molecular circuitry” or “gene networks.” A cell’s identity is the outcome of a complex web of interactions and is a dynamic, not a static condition. Regulatory factors are known to constantly cycle between the nucleus and the cytoplasm, affecting their own expression and other genes. So, a cell can be viewed as constantly updating its own program of gene expression. Where the regulatory cycle promotes continuation of a particular state, it is stable. And in the same way that a physical net can adopt different shapes when pulled or pushed, there are many stable patterns of gene expression and many differentiated states.
In a particular cell, some genes are highly expressed, some less so and others not at all. How that pattern is maintained and reliably passed onto daughter cells is incompletely understood. It is however known that chromatin, the complex of DNA wound around histone proteins, carries marks that relate to whether genes are active or inactive. These marks are “epigenetic” rather than genetic, in that they do not alter the actual DNA sequence. For example, DNA in and around inactive genes often carries methyl groups added onto the nucleotide cytosine. Active and inactive genes also show different chemical modifications to histones. This affects how tightly DNA is bound and how “open” and available it is to transcription factors.
When a nucleus of a differentiated cell is exposed to a foreign environment, for example when two cells are fused together, the regulatory processes are disrupted and the gene expression pattern alters accordingly. For example, a nucleus from a human liver cell can be induced to express muscle genes by fusion with a mouse muscle cell (Blau, Chiu, and Webster 1983) and the nuclei of several different somatic cells express embryonic genes when fused to ES cells (Do, Han, and Schöler 2006).
Nuclear transfer may be regarded as a more complete version of the same phenomenon. When a nucleus is transferred into an enucleated oocyte it undergoes comprehensive removal of the DNA methyl groups and major changes in histone modifications, thoroughly erasing its previous identity. Kevin Eggan and colleagues propose that the key to such successful reprogramming is the free availability of factors regulating gene transcription (Egli, Birkhoff, and Eggan 2008). These are normally associated with the DNA within the nucleus, but are released into the cytoplasm when the nucleus breaks down, ready to be distributed with the chromosomes into the two new nuclei. Unfertilized oocytes have an abundance of such free factors, being primed and ready to reprogram an incoming nucleus—the sperm head.
But what are the factors responsible for reprogramming a nucleus to an embryonic state? Unfortunately mammalian oocytes are tiny, do not propagate, and therefore difficult to analyze with current technology. So, researchers have turned to ES cells.
Direct reprogramming, a radical new approach
Years of intensive study have revealed much about the mechanisms that maintain ES cells in an undifferentiated state and trigger their differentiation. In 2006, this culminated in a major breakthrough. Shinya Yamanaka and colleagues of Kyoto University reasoned that regulatory factors known to be important in keeping ES cells undifferentiated would be good candidates for reprogramming factors. His group identified 24 regulatory genes and constructed viral vectors to transduce them individually into other cells. Different combinations of genes were then introduced into mouse fibroblasts and the cells selected for the expression of a gene characteristically expressed in ES cells. A set of four transcription factors: Sox-2, Oct-4, c-Myc, and Klf4, were found to convert the fibroblasts into something closely resembling ES cells, which they named induced pluripotent stem (iPS) cells (Takahashi and Yamanaka 2006) (Takahashi et al. 2007). Since their original report, Yamanaka and several other research groups have refined the technique and extended it to human cells (see Figure 4C). At the time of writing, the general opinion is that iPS cells and ES cells are essentially the same. However, these are early days and some critics have pointed out differences that may be significant (Liu 2008).
The discovery of a remarkably simple recipe to reprogram differentiated cells into an embryonic state has sparked an almost unprecedented frenzy of research activity around the world. Unlike nuclear transfer, there are no ethical concerns and the techniques are straightforward, opening the study of reprogramming to many laboratories. It has also caused some leading groups previously investigating therapeutic cloning to shift their research focus. The US Boston Globe of 1 August 2008 quotes Rudolf Jaenisch saying that the iPS approach “is so much easier, [with] so many fewer restrictions and problems—ethical as well as others, […] I think we’ll probably be moving in this direction.”
IPS cells are now such a fast moving area that this account will be out of date almost as soon as it is printed. But some important features are emerging as the story unfolds. At first it seemed that some cells could be reprogrammed and others not. But iPS cells have now been made from many cell types, such as mature B lymphocytes and pancreatic islet beta cells, demonstrating that it is not a quirk of some particular cell type, or an experimental artifact as some skeptics had claimed. Somewhat puzzlingly, different research groups are finding that widely different combinations and numbers of factors are effective. The underlying mechanisms are still obscure, but unlikely to remain a mystery for long. Bioinformatic analysis of whole constellations of genes is revealing the patterns of expression and the details of the regulatory networks that characterize ES and iPS cells and the events involved in direct reprogramming (Mikkelsen et al. 2008; Müller et al. 2008).
An immediate application of iPS cells is the study of degenerative diseases. Human iPS cells have already been isolated from patients with conditions such as motor neuron disease, Parkinson’s disease, Duchenne muscular dystrophy, and juvenile onset (type I) diabetes (Dimos et al. 2008; Park et al. 2008), and are being used to generate dishes of the affected cell type in the laboratory. The ready availability of disease-specific iPS cells will have a profound impact on the understanding and treatment of many serious disorders. It will allow the effect of environmental factors such as food additives, toxins, or pathogens on cell degeneration to be thoroughly examined in large-scale studies. Large numbers of drugs can also be screened to identify those that halt, slow, or reverse disease progression.
IPS cells have also raised considerable excitement as a source of replacement tissues without the need for human eggs or ES cells. In a proof-of-concept experiment, Rudolf Jaenisch successfully treated sickle-cell anemia in mice (Hanna et al. 2007). Skin cells from a mouse with sickle-cell anemia were converted to iPS cells and the genetic defect corrected by gene targeting. The iPS cells were induced to differentiate into blood stem cells and then transplanted into the mouse where they reduced anemia and increased survival.
However, it must be stressed that such iPS based cell therapies are still some way from the human clinic. Current methods of producing iPS cells involve potentially dangerous cancer genes, and alternatives must be found. Encouragingly, there are early reports that chemical agents can replace the need for some of the genes in the original recipe, and variations such as adding instructive RNA rather than genes are also being explored.
If iPS cells are identical to ES cells, they necessarily face the same issues regarding therapeutic use. Like ES cells, undifferentiated iPS cells can form tumors and so must be absolutely excluded from any therapeutic preparation. Methods must also be worked out to induce the differentiation of pure populations of therapeutic cell types. Differentiation conditions have been established for some, such as motor neurons, but procedures need to be worked out for many other potentially useful cells.
The two-year history of the iPS revolution has been breathtaking and has almost certainly made therapeutic cloning obsolete. But there are signs of yet another step change. While the oocyte was a black box that did not easily reveal its workings, the derivation of iPS cells has opened wide the study of reprogramming. Knowledge of the normal development of many cell types is also constantly improving and the role of key regulatory molecules becoming clear, allowing them to be used as “knobs and switches” to control cell identity. If the aim is to produce differentiated cells to order, why not do it directly without going through an embryonic intermediate? In a dramatic paper (Zhou et al. 2008) published in August 2008, Doug Melton and colleagues reported treating diabetic mice with three instructive genes carried in viral vectors. This induced some of the exocrine cells in the mouse pancreas, which normally secrete digestive enzymes, to convert directly to insulin producing beta cells without any intermediate formation of iPS, or other ES-like cells. Clearly this work is at a very early stage, but it has opened yet another route to the production of cells on demand (see Figure 4D). Most provocatively, because the work was carried out in mice not in culture there is now the prospect of patients requiring replacement therapy being able to ingest a cocktail of instructive factors designed to generate new cells within their own body with no need for transplantation. Could the regeneration of whole organs also now be on the horizon?
Concluding remarks
In a recent essay (Thomas 2007), John Meurig Thomas outlined the basic unpredictability of scientific progress and the tortuous paths that often lie between original research findings and the development of familiar modern devices and procedures. Famously, following their 1958 paper, Charles Townes and Arthur Schawlow foresaw no practical application for their invention, the optical laser.
Cloning was originally conceived to investigate the determination of cell identity and fate, but is now leading to the ability to change cell fate. Who knows what the most important legacy of Dolly will prove to be? After a little over eleven years, what is most evident is the intense attention she has brought to these questions, and the general sense of wide-open possibility and excitement. Many talented people have been attracted to this research and inspired to embark on projects that would have been inconceivable before 1997. We are optimistic that the current buzz of activity will bring significant advances in medicine and benefit to human health.
Bibliography
Beetschen, J. C., and J. L. Fischer. “Yves Delage (1854–1920) as a forerunner of modern nuclear transfer experiments.” Int. J. Dev. Biol. 48, 2004, 607–612.
Blau, H. M., C. P. Chiu, and C. Webster. “Cytoplasmic activation of human nuclear genes in stable heterocaryons.” Cell 32, 1983, 1171–1180.
Briggs, R., and T. J. King. “Transplantation of living nuclei from blastula cells into enucleated frogs’ eggs.” Proc. Natl. Acad. Sci. USA. 38, 1952, 455–463.
Campbell, K. H., J. Mcwhir, W. A. Ritchie, and I. Wilmut. “Sheep cloned by nuclear transfer from a cultured cell line.” Nature 380, 1996 64–66.
Dimos, J. T., K. T. Rodolfa, K. K. Niakan, L. M. Weisenthal, H. Mitsumoto, W. Chung, G. F. Croft, et al. “Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons.” Science 321, 2008, 1218–1221.
Do, J. T., D. W. Han, and H. R. Schöler. “Reprogramming somatic gene activity by fusion with pluripotent cells.” Stem Cell Rev. 2, 2006, 257–264.
Driesch H. “Entwicklungsmechanisme Studien. I. Der Werth der beiden ersten Furchungszellen in der Echinodermentwicklung. Experimentelle Erzeugen von Theil und Doppelbildung.” Zeit. für wiss. Zool 53: 160–178, 1892, 183–184.
Egli, D., G. Birkhoff, and K. Eggan. “Mediators of reprogramming: transcription factors and transitions through mitosis.” Nat. Rev. Mol. Cell. Biol. 9, 2008, 505–516.
Frese, K. K., and D. A. Tuveson. “Maximizing mouse cancer models.” Nat. Rev. Cancer 7, 2007, 645–658.
Gurdon, J. B. “The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles.” J. Embryol. Exp. Morphol. 10, 1962, 622–640.
Hanna, J. M. Wernig, S. Markoulaki, C. W. Sun, A. Meissner, J. P. Cassady, C. Beard, et al. “Treatment of sickle cell anemia mouse model with iPS cells generated from autologous skin.” Science 318, 2007, 1920–1923.
Hochedlinger, K., and R. Jaenisch. “Monoclonal mice generated by nuclear transfer from mature B and T donor cells.” Nature 415, 2002, 1035–1038.
Illmensee, K., and P. C. Hoppe. “Nuclear transplantation in Mus musculus: developmental potential of nuclei from preimplantation embryos.” Cell 23, 1981, 9–18.
Kuroiwa, Y., P. Kasinathan, H. Matsushita, J. Sathiyaselan, E. J. Sullivan, M. Kakitani, K. Tomizuka, I. Ishida, and J. M. Robl. “Sequential targeting of the genes encoding immunoglobulin-mu and prion protein in cattle.” Nat. Genet. 36, 2004, 775–780.
Liu, S.V. “iPS cells: a more critical review.” Stem Cells Dev. 17, 2008, 391–397.
Lysenko, T. D. Soviet Biology: Report and concluding remarks to the 1948 session of the Lenin Academy of Agricultural Sciences. (English ed.) London: Birch Books, 1948. Online version: www.marxists.org/reference/archive/lysenko/works/1940s/report.htm
Mcgrath, J., and D. Solter. “Inability of mouse blastomere nuclei transferred to enucleated zygotes to support development in vitro.” Science 226, 1984, 1317–1319.
Maher, B. “Egg shortage hits race to clone human stem cells.” Nature 453, 2008, 828–829.
Mikkelsen, T. S., J. Hanna, X. Zhang, M. Ku, M. Wernig, P. Schorderet, B. E. Bernstein, R. Jaenisch, E. S. Lander, and A. Meissner. “Dissecting direct reprogramming through integrative genomic analysis.” Nature, 454, 2008, 49–55.
Morgan, T.H., A. H. Sturtevant, H. J. Muller, and C. B. Bridges. The Mechanism of Mendelian Heredity. New York: Henry Holt and Co., 1915.
Müller, F. J., L. C. Laurent, D. Kostka, I. Ulitsky, R. Williams, C. Lu, I. H. Park, et al. “Regulatory networks define phenotypic classes of human stem cell lines.” Nature, 455, 2008 401–405.
Park, I. H., N. Arora, H. Huo, N. Maherali, T. Ahfeldt, A. Shimamura, M. W. Lensch, C. Cowan, K. Hochedlinger, and G. Q. Daley. “Disease-specific induced pluripotent stem cells.” Cell 134, 2007, 877–886.
Rogers, C. S., Y. Hao, T. Rokhlina, M. Samuel, D. A. Stoltz, Y. Li, E. Petroff, et al. “Production of CFTR-null and CFTR-DeltaF508 heterozygous pigs by adeno-associated virus-mediated gene targeting and somatic cell nuclear transfer.” J. Clin. Invest. 118, 2008, 1571–1577.
Schnieke, A. E, A. J. Kind, W. A. Ritchie, K. Mycock, A. R. Scott, M. Ritchie, I. Wilmut, A. Colman, and K. H. S. Campbell. “Human factor IX transgenic sheep produced by transfer of nuclei from transfected fetal fibroblasts.” Science 278, 1997, 2130–2133.
—. “Human factor IX transgenic sheep produced by transfer of nuclei from transfected fetal fibroblasts.” Science 278, 1997a, 2130–2133.
—,“Production of gene-targeted sheep by nuclear transfer from cultured somatic cells.” Nature 405, 1997b, 1066–1069.
Signer, E. N., Y. E. Dubrova, A. J. Jeffreys, C. Wilde, L. M. Finch, M. Wells, and M. Peaker. “DNA fingerprinting Dolly.” Nature 394, 1998, 329–330.
Silva, J., and A. Smith. “Capturing pluripotency.” Cell 132, 2008, 532–6.
Spemann, H. “Die Entwicklung seitlicher und dorso-ventraler Keimhälften bei verzögerter Kernversorgung.” Zeit. für wiss Zool 132, 1928, 105–134.
—, Experimentelle Beiträge zu einer Theorie der Entwicklung. Berlin: Springer, 1936. (English ed., Embryonic development and induction, 1938.)
Takahashi, K., K. Tanabe, M. Ohnuki, M. Narita, T. Ichisaka, K. Tomoda, and S. Yamanaka. “Induction of pluripotent stem cells from adult human fibroblasts by defined factors.” Cell 131, 2007, 861–872.
Takahashi, K. and Yamanaka, S. “Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors.” Cell 126, 2006, 663-76.
Thomas, J. M. “Unpredictability and chance in scientific progress.” Progress in Informatics 4, 2007, 1–4.
Thomson, J. A., J. Itskovitz-Eldor, S. S. Shapiro, M. A. Waknitz, J. J. Swiergiel, V. S. Marshall, and J. M. Jones. “Embryonic stem cell lines derived from human blastocysts.” Science 282, 1998, 1145–1147.
Wakayama, T., A. C. Perry, M. Zuccotti, K. R. Johnson, and R. Yanagimachi. “Full-term development of mice from enucleated oocytes injected with cumulus cell nuclei.” Nature 394, 1998, 369–374.
Weismann, A. Das Keimplasma. Eine Theorie der Vererbung. Jena: Gustav Fischer, 1892.
Willadsen, S. M. “Nuclear transplantation in sheep embryos.” Nature 320, 1986, 63–65.
Wilmut, I., A. E. Schnieke, J. Mcwhir, A. J. Kind, and K. H. Campbell. “Viable offspring derived from fetal and adult mammalian cells.” Nature 385, 1997, 810–813.
Woods, G. L., K. L. White, D. K Vanderwall, G. P. Li, K. I. Aston, T. D. Bunch, L. N. Meerdo, and B. J. Pate. “A mule cloned from fetal cells by nuclear transfer.” Science 301, 2003, 1063.
Zhou, Q. J. Brown, A. Kanarek, J. Rajagopal, and D.A. Melton. “In vivo reprogramming of adult pancreatic exocrine cells to beta-cells.” Nature, Aug 27, 2008. [Epub ahead of print.]
Comments on this publication