Background: Why Is Aging Still Resisting Medical Intervention?
The goal of bringing aging under comprehensive medical control is probably humanity’s oldest dream—and it is certainly humanity’s foremost problem today. However, our progress toward it is lamentably slight. The history of our attempts to control aging can be summarized as a sequence of mis-steps: of moisguided and (in hindsight, anyway) mistaken approaches that never had a chance of succeeding. They can each be summarized in a single word: disease, design, and deprivation. And the worst of it is that they have not even been sequential: the earlier ones have survived the arrival of the later ones.

The “aging as disease” false dawn, otherwise known as geriatric medicine, rests on the assumption that the diseases of old age are inherently amenable to the same kind of medical assault as the most prevalent diseases of youth, that is, infections. It arises from the observation that different people get different diseases in old age, just as different people get different infections, and the inference that the same kind of strategy against the latter will work against the former—which means, attacking the symptoms of the disease. How crazy is that, when the ailments of old age are consequences of the accumulation of self-inflicted damage in the body, while infections are from elsewhere and can therefore be banished entirely? But that has not stopped, and still does not stop, humanity from expending billions upon billions of dollars on research aimed at achieving this patently quixotic goal. Man rightly prefers it to be true, and evidently also prefers to believe, that these diseases are “curable”—and no amount of logic seems to be able to change that.
The “aging as design” false dawn emerged a century or so ago, perhaps partly as a result of a few people’s recognition of the above argument. It began with August Weismann’s proposal that aging serves an evolutionary purpose—a seductive idea that survived in its initial form for several decades despite being inherently circular (essentially, Weissman was saying that aging is selected because it exists, as well as the other way around), and still survives among a minority of the field in a more sophisticated form (typically couched in terms of evolutionary nimbleness, or “evolvability”). But that has not been its main legacy. Rather, it gave rise in the early twentieth century to an approach that does not rely on the idea that there exist genes whose selected function is to hasten the organism’s decline in function, but instead on the idea that the genes determining the variation between species (and the lesser, but still non-negligible, variation between individuals of the same species) are rather few in number, and thus that it is realistic to seek to tweak those of a given species (such as Homo sapiens) so as to extend its healthy lifespan. It is easy to see how the latter idea is inspired by the former, but unfortunately it is not reliant on the former (because it can equally hold if the genes in question are anti-aging genes rather than pro-aging ones), and therefore its rejection by most gerontologists did not follow from that of “programmed aging.”
Again, though, we must ask the key question: does the “aging as design” basis for the pursuit of medical postponement of age-related ill health actually make sense? Is it even remotely compatible with what we know about aging? And again, the painfully obvious answer is no. Precisely for the same reasons that we know aging is not programmed, we also know that it is inherently chaotic and multifactorial. Different types of damage are created by different mechanisms, and different genetic pathways are devoted to slowing those mechanisms and thereby postponing the age at which the corresponding type of damage reaches pathogenic abundance. And, since selective pressure is imposed upon those pathways by a common mechanism, namely the relationship between an individual’s function and its ability to contribute to the next generation, they are inexorably destined (except in special cases that I have discussed under the phenomenon of “protagonistic pleiotropy”) to gravitate to levels of efficacy that result in the corresponding pathologies emerging at about the same age. Thus, there is no possibility whatsoever that substantial slowing of the accumulation of all of these types of damage could be simultaneously effected by the tweaking of a small number of genes. And yet, just as with geriatric medicine, faith in the existence of some elusive “magic bullet” has persisted in the minds of a depressing number of biologists.
But is it that bad? Do biologists whose main area is the biology of aging think this way? Not entirely. For sure, many biogerontologists select their research direction in a manner suggestive of such an error, but we must remember that (as in any field of science) most researchers are driven by curiosity rather than by translational potential, and I would be the last to suggest that curiosity driven research is worthless. So I do not condemn those researchers in the way that I condemn geriatrics. And the biogerontologists who do think substantially in terms of translational potential came, by and large, to recognize the above logic quite well by the 1960s or so.
So far so good—in a way. During the 1970s and 1980s, there was a depressing downside to this, namely that almost all translationally motivated gerontologists responded to this realization by becoming the other sort of gerontologist, the curiosity driven type, to such an extent that it became impossible even to entertain (publicly, or in grant applications, etc.) the possibility of eventual postponement of aging without fatally damaging one’s fundability. But at least they were being realistic.
Until… the third false dawn. By “deprivation” I refer, as I hope you have guessed, to calorie restriction (CR), an intervention that was shown as early as the 1930s to extend the lives of mice and rats by as much as fifty percent. It had also been shown to have a proportionally (and anatomically) much more dramatic effect in nematodes, which enter a radically different developmental pathway when starved at a particular stage in their growth. In hindsight it is therefore curious that, given these well-known facts, the biogerontology community was at first highly skeptical of and then highly energized by reports in the late 1980s and early 1990s that the same effect, with only slightly lesser magnitude, could be achieved in nematodes by genetic means. But whatever the history, the fact is that “energized” is a decided understatement.
To this day, biomedical gerontology research is hugely dominated by the quest for better ways to emulate the effect of calorie restriction by genetic—or, more recently, pharmacological—means.
Why is this a third false dawn? Because its true biomedical potential is, and has long been, obviously almost nil. The reason this is so obvious arises from the combination of two points. First, virtually without exception (and the one “known” exception is in fact probably incorrect, though worthy of further investigation), no genetic or pharmacological intervention in any species has ever exceeded the performance of CR itself in the same species when initiated at the same age—which is exactly what everyone should have expected, since the life extension conferred by CR and by its emulation must be mediated by the same genetic pathways, pathways whose effect just is what it is. And second, that performance of CR itself varies inversely with the non-CR longevity of the species: longer-lived species derive much less benefit as a proportion of their lifespan, and in fact not much more benefit in absolute time. This too should have been expected, since the selective pressure giving rise to the pathways that mediate the response to CR arises from the frequency of famines, which is independent of the lifespan of the organisms experiencing those famines. Since long famines are rarer than short ones, the prediction is clear. And adding this all up, the conclusion is equally clear: emulation of CR will not significantly postpone human aging. But would you know this from a perusal of the grant applications currently funded by the US National Institute on Aging, or its counterparts worldwide? Not as such. If we exclude, as discussed above, the grants whose focus is purely to extend our understanding of aging rather than to do anything about it, the overwhelming majority concern one or another approach to emulating CR. Just as with the other false dawns, this concept is clung to in the face of the utterly iron-clad logic to the contrary, simply because its proponents have sunk so much cost in terms of intellectual investment in it that their only route to self-preservation is to keep on approving each other’s grant applications.
The Fourth Way: Damage Repair
But this century, step by painfully small step, things are changing. In the remainder of this essay, I shall describe the progress that has so far occurred in implementing the approach to combating aging that I first introduced about fifteen years ago, under the name “Strategies for Engineered Negligible Senescence” (SENS). Much of this progress has occurred under the leadership of the biomedical research charity created for that purpose, SENS Research Foundation.
It may seem curious that a charity dedicated, in part, to improving public awareness employs as its name a phrase as technical as this. Indeed, we have recently introduced the more lay-friendly term “rejuvenation biotechnology” to encompass the technologies required to implement SENS. “Rejuvenation,” however, is not an obviously quantifiable concept, and the original phrase remains the most accurate description of our goal.
“Senescence,” here, refers to the actuarial phenomenon—the trend that individuals within a population suffer from an increasing morbidity and mortality rate in (typically exponential) relation to their chronological age.
“Negligible” is used in a statistical sense: we consider a level of senescence negligible if no age-related contribution to mortality is statistically demonstrable within a population, given the “background noise” of age-independent mortality (such as unfortunate encounters with motor vehicles). We accept that this represents a moving target; a non-negligible level of senescence might be observed in the same population after improvements in available data or analytical methods, requiring further iterations of the therapeutic platform. In fact, several such cycles seem likely.
Finally, by “Engineered,” we indicate that this state is achieved by the deliberate application of biomedical therapies, and is not the normal situation (as observed in, for example, Hydra, where continual regeneration appears to result in a lack of any correlation between age and mortality rate—although the indefinite maintenance of reproductive potential is more controversial).
Our goal is thus unambiguously defined; we seek methods to convert a population experiencing a non-negligible level of senescence into one experiencing a negligible level.
To most readers, this goal—the comprehensive elimination of age-related degeneration—will at first seem outlandish. Certainly such an accomplishment has been an aspiration of humanity for at least the whole of recorded history, with little result. However, there are good reasons to think that it could be more easily achievable than “just” slowing the process of aging such that the age-related increase in mortality is reduced and the average lifespan modestly extended. In this essay, I outline those reasons.
To see how the goal of negligible senescence could be “engineered,” it is useful to consider a situation in which human ingenuity and perseverance has already achieved an analogous result. Motor vehicles experience a process of wear-and-tear essentially similar to organismal aging; the paint flakes, windowpanes chip, rust infiltrates the pipework, and so forth. Nonetheless, as vintage car owners will attest, it is entirely possible to keep one functional and even pristine for an essentially indefinite period. Critically, this is achieved not by preventing the wear—we can see, instinctively, that doing so perfectly is impossible—but by repairing the damage that does occur at a rate sufficient to ensure that the function of the machine is never irretrievably compromised.
Of course, the analogy is inexact; human bodies are far more complex than cars, and we are still a long way from understanding how they are constructed and the processes that contribute to wearing them down. Fortunately, a closer look at precisely how growing older leads to debility reveals that our ignorance need not be showstopping.
Aging can be characterized as a three-stage process. In the first stage, metabolic processes essential to life produce toxins—an unavoidable side effect of handling reactive chemicals. Secondly, a small amount of the damage caused by these toxins cannot be removed by the body’s endogenous repair systems, and consequently accumulates over time. In the third stage, the accumulation of damage drives age-related pathology, either directly—by interfering with normal metabolism—or indirectly, often where a normally benevolent repair process causes collateral damage due to maladaptive overstimulation.
This model (“metabolism causes damage causes pathology”) allows us to clarify, considerably, the requirements for successful intervention in aging. Unlike the dynamic processes of metabolism and pathology—which we understand so poorly, and where a single intervention invariably triggers a cascade of knock-on effects—accumulated damage represents a relatively stationary target. That is to say, it may not be clear whether a given type of damage is pathological (on balance), but its absence from healthy twenty-year-olds indicates that it is not required for healthy life. Conversely it is clear that the total ensemble of types of damage is pathological, since fifty-year-olds have considerably less time to live than twenty-year-olds, and the only static difference between the two groups is the amount of accumulated damage present. (There are dynamic differences, where the body reacts maladaptively to the presence of damage, but the profoundly homeostatic nature of physiology gives us confidence that this will self-correct once the stimulus for the maladaptive response is removed.)
Accepting the implications of this model (which replicates aging-induced mortality curves reassuringly well) leads us to the SENS approach; by identifying and repairing all of the damage accumulated during aging, we can restore the body to a youthful state. Consequently, its dynamic metabolic processes will revert to their own norms, and the risk of mortality will be no higher than in any other equivalently “youthful” individual—whether they have actually lived for twenty years or 120. Furthermore—so long as our inventory of damage classes is sufficiently comprehensive—we can repeat this effort on a regular basis, and thus remain indefinitely below the threshold of pathology. Crucially, we can do this without a comprehensive understanding of the complex metabolic processes giving rise to damage, nor of those leading from damage to pathology. We need only an inventory of the types of damage which exist, which can be obtained directly by comparison of older and younger individuals.
What Is the Damage of Aging?
Fortunately, it seems that all aging-related damage known to accumulate in the human body can be classified into just seven clearly defined categories. For each category, the SENS platform incorporates a mechanism for repairing the damage, using either existing biotechnology or readily foreseeable extensions thereof. I now briefly summarize these categories, and the associated therapeutic options for their resolution.
Cell Loss
The most straightforward damage category is the loss (death without replacement) of cells, either as a result of chronic injury or acute trauma, mediated by apoptosis, by autophagic cell death, and/or by necrosis. Such losses lead to tissue atrophy, compromising the function of the organs involved; examples include Parkinson’s disease, sarcopenia, autoimmune diabetes, and presbycusis. Therapy involves the introduction of stem or progenitor cells (and/or stimulation of the proliferation of endogenous progenitors), either systemically with the assistance of appropriate targeting mechanisms, or locally via a tissue-engineered construct.
Cell Death-Resistance
Conversely, the accumulation of excessive numbers of cells refractory to normal homeostatic apoptosis can also be harmful. The most obvious example in the context of Western societies is obesity, but there are more subtle manifestations—the imbalance between anergic and naïve T-lymphocytes that characterizes immunosenescence being a prime example. Treatment is in this case conceptually straightforward; identify biomarkers that selectively target the undesirable cells, and deliver cytotoxic drugs or viruses to ensure their destruction.
Cell Over-Proliferation
The last (but definitely not least) “cellular” category deals with damage to the genomic DNA, in the form both of mutation (changes to the base-pair sequence) and epimutation (changes to the moieties that decorate the DNA molecule and influence its transcription). Fortunately, this is one area in which evolution has done most of the hard work for us. Since the emergence of vertebrates (at least), the most serious problem originating from mutation has been cancer, which has the capacity to kill an organism if one single cell acquires enough mutational load—whereas any other mutation can in general become lethal only if it afflicts a substantial proportion of the cells in a given tissue. The proofreading mechanisms evolved to prevent cancer are, as a result, more than sufficient to keep non-cancerous mutations under control. Thus, this strictly molecular category of damage is in fact best treated as a cellular one.
Of course, cancer itself remains a very real issue. The SENS solution—WILT, an acronym for “Whole-body Interdiction of Lengthening of Telomeres”—is the only genuinely universal anti-cancer therapy to have been proposed. WILT is a two-pronged approach that involves the deletion (not merely the inactivation) of the telomerase and ALT (alternative lengthening of telomeres) genes throughout the body, preventing any newly formed cancer from growing large enough to become life-threatening, accompanied by (approximately) decadal re-seeding of our rapidly renewing tissues’ stem cell reserves using cells whose telomeres have been lengthened ex vivo. As ambitious as this is, WILT’s comprehensiveness if it can be implemented is not disputed, so it remains a “therapy of last resort” that we would be unwise to neglect until and unless simpler therapies of comparable efficacy are developed.
The remaining four categories of damage are molecular, rather than cellular: two are intracellular and two extracellular.
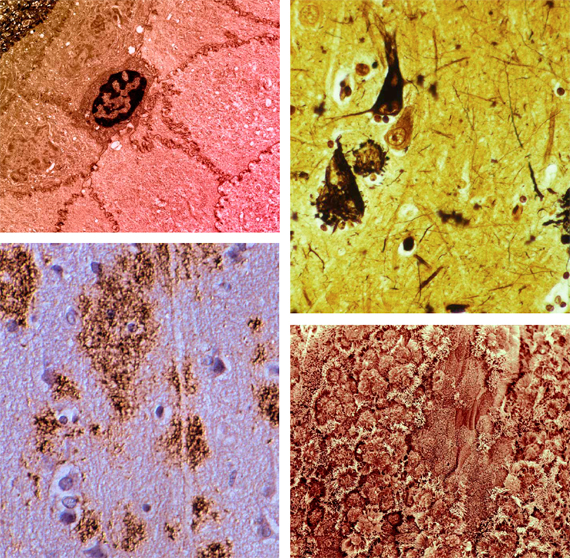
Intracellular “Junk”
While cells are generally very efficient in detecting (particularly via the unfolded protein response) and recycling damaged or surplus biomolecules, there are some exceptions—substances which are genuinely refractory to degradation or excretion. The resulting accumulation of “junk” compromises the cell’s metabolism—at best impairing its normal function, and at worst leading to cell death. The accumulation of lipoproteins in atherosclerotic plaques and the phototoxicity of A2E that drives dry macular degeneration are two of the best-studied examples of this mechanism, which may be treated by augmenting the native degradation machinery by introducing novel enzymes capable of tackling the most recalcitrant targets. Such enzymes typically need to be introduced into the lysosome, cells’ “garbage disposal machinery of last resort.”
Extracellular “Junk”
Not all waste products accumulate within the cell; the infamous amyloid plaques of Alzheimer’s disease, as well as the amyloids associated with type II diabetes and systemic amyloidosis, accumulate in the extracellular space. Fortunately, the body already possesses an excellent mechanism for removing undesirable substrates from this compartment—the phagocytic cells of the immune system—and thus the elimination of this junk is essentially a problem of developing an effective and safe vaccine. (In a few cases, it may be necessary to apply the principles of the previous category—lysosomal enhancement—to ensure that the immune cells are able to fully degrade the substances they consume.)
Tissue Stiffening
Protein comprises the bulk of the extracellular matrix; in many cases the proteins involved are laid down in early life and then recycled very slowly, if at all, in adulthood. The function of the tissues supported by such proteins—such as the elasticity of the artery wall—is dependent on their maintaining the correct molecular structure. As the years pass that structure becomes progressively compromised, primarily due to crosslinking induced by reactive molecular species, especially a network of reactions initiated by circulating sugars and collectively termed “glycation.” In the arteries, this stiffening leads to increasing blood pressure and all its downstream consequences. Fortuitously, such crosslinks are chemically distinct from any that are enzymatically derived, and the therapeutic strategy is thus to develop drugs or enzymes capable of cleaving these crosslinks to restore the original biophysical function.
Mitochondrial Defects
Mitochondrial DNA (mtDNA) is uniquely vulnerable, being in close proximity to an intense source of reactive species (the respiratory chain) while lacking many of the sophisticated repair mechanisms available to nuclear DNA. There is a well-established accumulation of damage to mtDNA associated with aging—linked, as one would expect where such a vital cellular component is involved, to a very broad range of degenerative conditions. However, of the thousand or so protein components of a mature mitochondrion, only thirteen are actually encoded in the mtDNA—the remaining genes having moved over evolutionary time to the nuclear DNA, from where their products are efficiently translocated back to the mitochondrion. The solution in this case is thus not to repair damage to the remaining genes, but rather to introduce translocation-ready variants of them into the nuclear genome—a technique termed allotopic expression—obviating the issue of maintaining the mtDNA itself. We should emphasize that the above seven-category formulation is descriptive, not prescriptive. However, the fact that no new categories of damage have been discovered since 1982, despite the dramatic progress in bioanalytical techniques since that time, suggests that any other categories accumulate too slowly to have any bearing on health within the contemporary human lifespan.
Recent Progress
Since the SENS approach was first proposed in 2002, marked progress has been made in every relevant area of research. For the remainder of this essay, I will highlight a few of the most exciting and recent discoveries, and explain how they contribute to the development of effective rejuvenation biotechnologies.
Why Recycling Will Save More Than Just the Planet
Some of the best recent evidence in favor of a strategy based on the removal of accumulated damage has come from work on autophagy—the process by which cells naturally recycle proteins, lipids, and even whole organelles. In particular, chaperone-mediated autophagy (CMA), a process that mediates the translocation of proteins bearing a specific motif (either inherently, or by virtue of unfolding or post-translational modification) to the lysosome for degradation, has been shown to decrease with age due to reduced availability of the LAMP (lysosome-associated membrane protein)-2A receptor.
Mice engineered with inducible LAMP-2A in the liver (where the aging-related dysfunction of autophagy is well characterized) show markedly reduced levels of oxidized protein accumulation and structural degeneration at twenty-two months, with hepatocyte function closer to that of young (six months) mice than to wild-type littermates. Intriguingly, the transgenic mice also exhibited enhanced rates of macroautophagy and ubiquitin-proteasome function, implying a positive feedback between damage repair systems.
Enhancing the trafficking of damaged biomolecules is, of course, only one way to increase their rate of recycling. In very recent work in mouse models of Alzheimer’s disease, up-regulation of the lysosomal protease cathepsin B has been shown to result in rapid and dramatic clearance of Aβ(1-42)—followed by substantial recovery of behavioral deficits in multiple tests.
Tools, Borrowed and Found
Promising though approaches founded on the enhancement of existing recycling machinery are, there are some aging-related waste products which appear to be entirely refractory to degradation by the cell’s normal lytic enzymes. These targets—including the toxic bisretinoid A2E, some forms of oxidized cholesterol, and others—require a qualitatively different method of removal.
One early insight in the development of the SENS platform was the observation that many such substances, while resistant to degradation by human cells, do not accumulate even in soils greatly enriched in human remains. Therefore, it was reasoned that some organism must exist that was competent to metabolize them. Bacterial species with just such a capacity were indeed rapidly identified, and the process of determining the enzymes involved and adapting them for therapeutic use forms one major branch of the work being undertaken at SENS Foundation’s Research Center.
A more recent development in this area was the discovery of catalytic antibodies (those with direct hydrolytic activity against a particular antigen, presumably due to superior affinity for the transition as compared to the ground state) against amyloid-β. Direct immunotherapy against amyloid deposits hit a stumbling block in clinical trials due to the damaging side effects of inflammation in the brain—a problem that is particularly pronounced in patients with advanced disease, and hence high amyloid load. Catalytic immunotherapy, on the other hand, avoids this concern as the hydrolyzed protein is not sequestered in a form prone to generating an immune response. Simultaneously, by virtue of the high specificity of the antibody, the strategy avoids the off-target effects seen with conventional enzyme approaches to amyloid hydrolysis (such as neprilysin).
The mechanism by which therapeutic antibodies of any kind cross the blood-brain barrier is an area of active enquiry. Although penetrance of immunoglobulin class G (IgG) is very poor, IgM and Fab fragments have been shown to cross the barrier much more effectively, and in any case a poorly penetrant but otherwise effective agent might be assisted in entry by manipulating the process of receptor-mediated transport used by substances such as insulin and transferrin.
SENS Foundation is sponsoring a project to determine whether catalytic antibody technology can be effectively applied to cardiac amyloidoses—which affect heart function in as many as twenty-five percent of those over the age of eighty, and appear to be the most common cause of death at the extremes of current human lifespan.
When Nature Does Not Provide
In dealing with recalcitrant waste substances, or crosslinks between matrix proteins, the challenge is generally to identify an enzyme (or small molecule) capable of catalyzing the degradation of a particular species into one more susceptible to the body’s native recycling machinery. The gold standard for accomplishing this goal within the pharmaceutical industry is high-throughput screening—testing enormous arrays of candidates in the hope of identifying one or more with the desired activity.
Effective as high-throughput screens can be, they are essentially a biochemical sweepstake. Consequently, it is occasionally argued that types of damage may exist which cannot be cleared simply because the agent required to repair them is absent from all existing chemical libraries—or has never been synthesized at all. Is this a problem for the comprehensiveness of the SENS approach?
While the field is still just in its infancy, the computational design (and optimization by directed evolution) of enzymes able to catalyze an arbitrary reaction by multiple orders of magnitude has recently been shown to be feasible. The applications of this approach in industry—let alone in medicine—are so self-evident that its refinement seems inevitable. Thus, we feel confident that no specific form of chemical damage will be forever irreparable.
Spare Parts and Overnight Hearts
While in situ repair at the cellular and molecular level is the most ideal solution to aging-related damage, we do not anticipate that it will always be possible—particularly while rejuvenation biotechnology is in its infancy. In some cases, the damage will be too extensive or heterogeneous for repair to be feasible; in others, the exact molecular nature of the damage may be so unclear as to make a direct attack difficult to formulate.
In such cases, a reasonable interim solution originates from the field of tissue engineering; instead of rejuvenating the existing organ, we would seek to replace it with an entirely new one grown in vitro. This concept is hardly new, but has tended to suffer from three major impediments; firstly the problem of achieving adequate vascularization to support the growing tissue; secondly (and particularly where replacement is urgent, as is usually the case) the prolonged periods of proliferative growth necessary to approach the size and function of an adult organ; and thirdly the requirement, when allogeneic cells or tissues are used, for lifelong immunosuppression.
Two new methods for organ synthesis are now showing considerable promise in overcoming these limitations. In the first—decellularization—donor tissue is treated with a detergent and enzyme mixture until the original cells and donor antigens have been stripped away, leaving a vacant, non-immunogenic scaffold. This scaffold is then incubated for several days with patient-derived stem/progenitor cells, which repopulate it and eventually reconstitute normal tissue function. Notably, although the scaffold-preparation step requires weeks, it can be conducted without knowledge of the recipient; only the much briefer repopulation stage is patient-specific. Thus far demonstrated in the successful transplantation of a large tracheal graft, this technique seems certain to become more widely employed in the near future.
The second novel technique is bioprinting—the preparation of a graft of almost any desired shape by depositing cells in a manner similar to the operation of an inkjet printer. Bioprinting eliminates the need for a donor scaffold, and thus should eventually prove more practical in general use. Although it is at a very early stage in terms of therapeutic application, the prospect of a relatively unlimited supply of highly similar organs, ideal for drug safety trials, is driving substantial preclinical investment.
An Ounce of Prevention
We have already introduced the concept of allotopic expression—transferring the remaining thirteen protein-coding genes of the mtDNA to the nuclear genome, where they will benefit from its superior maintenance mechanisms and isolation from the most dangerous sources of reactive species. Of course, the rather concise description previously offered glosses over the technical challenges involved.
The proteins in question are among the most hydrophobic of those found in the mitochondria; consequently, once folded after synthesis in the cytoplasm, they are the most difficult to unfold again, a prerequisite for translocation across the inner mitochondrial membrane. When SENS was first proposed, two complementary strategies were put forward to facilitate their import. The first was the substitution of highly hydrophobic amino acid residues with less hydrophobic relatives; the second involved the introduction of self-splicing inteins into the most tightly folded regions, preventing their folding entirely until after translocation. Of course, the former strategy seemed worryingly likely to impair the protein’s function (even when point mutations known to be non-pathogenic in isolation were employed), and the second relied on a new and largely untested biotechnology.
In 2006, a French group working on heritable mitochondriopathies demonstrated that by introducing a targeting sequence into the 3’ untranslated region of the mRNA for ATP6 (a subunit of ATP synthase, and one of the thirteen mtDNA-encoded proteins) it was possible to localize the mRNA to the mitochondrial surface. The localization achieved is in fact so tight that the import becomes “co-translational”—the amino acid chain is fed through the import machinery as it is synthesized, before folding, obviating the entire issue of its hydrophobicity. The mechanism was subsequently demonstrated to also be effective for the ND1 and ND4 genes. With the blessing of the technique’s originators (who are pursuing clinical translation of their original results), staff at the SENS Foundation Research Center are working to confirm that the same methodology can be applied to the ten remaining mtDNA-encoded proteins.
Conclusion
SENS is a hugely radical departure from prior themes of biomedical gerontology, involving the bona fide reversal of aging rather than its mere retardation. By virtue of a painstaking process of mutual education between the fields of biogerontology and regenerative medicine, it has now risen to the status of an acknowledged viable option for the eventual medical control of aging. I believe that its credibility will continue to rise as the underlying technology of regenerative medicine progresses.
Comments on this publication